
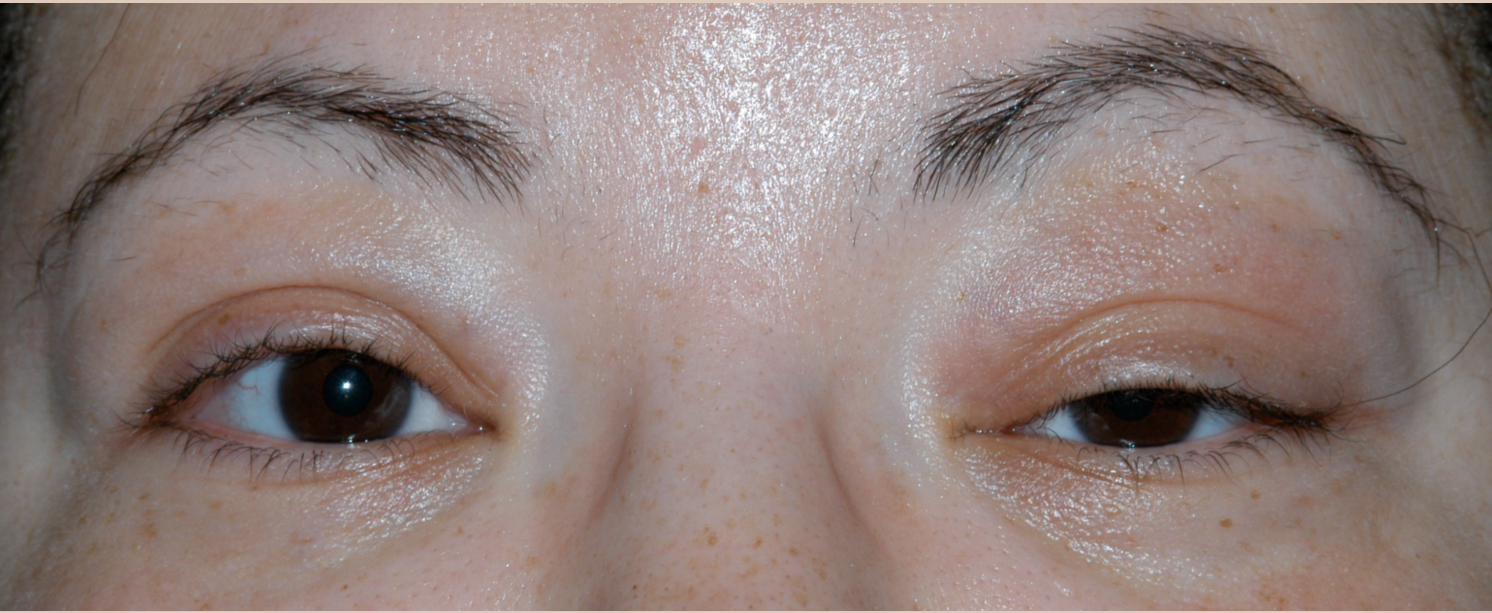
Chief Complaint: Left upper eyelid swelling
The patient was a 39-year-old female who presented with left upper eyelid swelling that started 4 months ago. She denied pain but felt a sense of fullness over the left eye. Occasionally her eyelid would itch or burn and have some associated mattering. She denied visual deficits or double vision. She had no history of facial or ocular trauma, sinus or skin infections, or ocular surgeries in the past.
Medical History: Type 2 diabetes mellitus for 9 years, myopia, polycystic ovarian syndrome
Past Surgical History: None
Social History: No tobacco or alcohol use
Family History: Cancer, kidney disease, and stroke (all in her mother); hypertension (mother and father), diabetes (material aunt and uncle, paternal grandparents), arthritis (maternal grandmother), lupus erythematosus (mother), cataracts
Medications: Metformin, doxycycline, levonorgestrel-ethinyl estradiol
No known drug allergies
Review of Systems: No constitutional symptoms, no cough, shortness of breath, chest pain or neurological symptoms
Visual acuity with correction
Intraocular pressure by applanation
Pupils: 4 → 2 mm, no relative afferent pupillary defect in either eye
Confrontation visual fields: Full OU
Extraocular motility: Full OU
Slit lamp exam: Meibomian gland dysfunction OU, normal exam without anterior segment inflammation
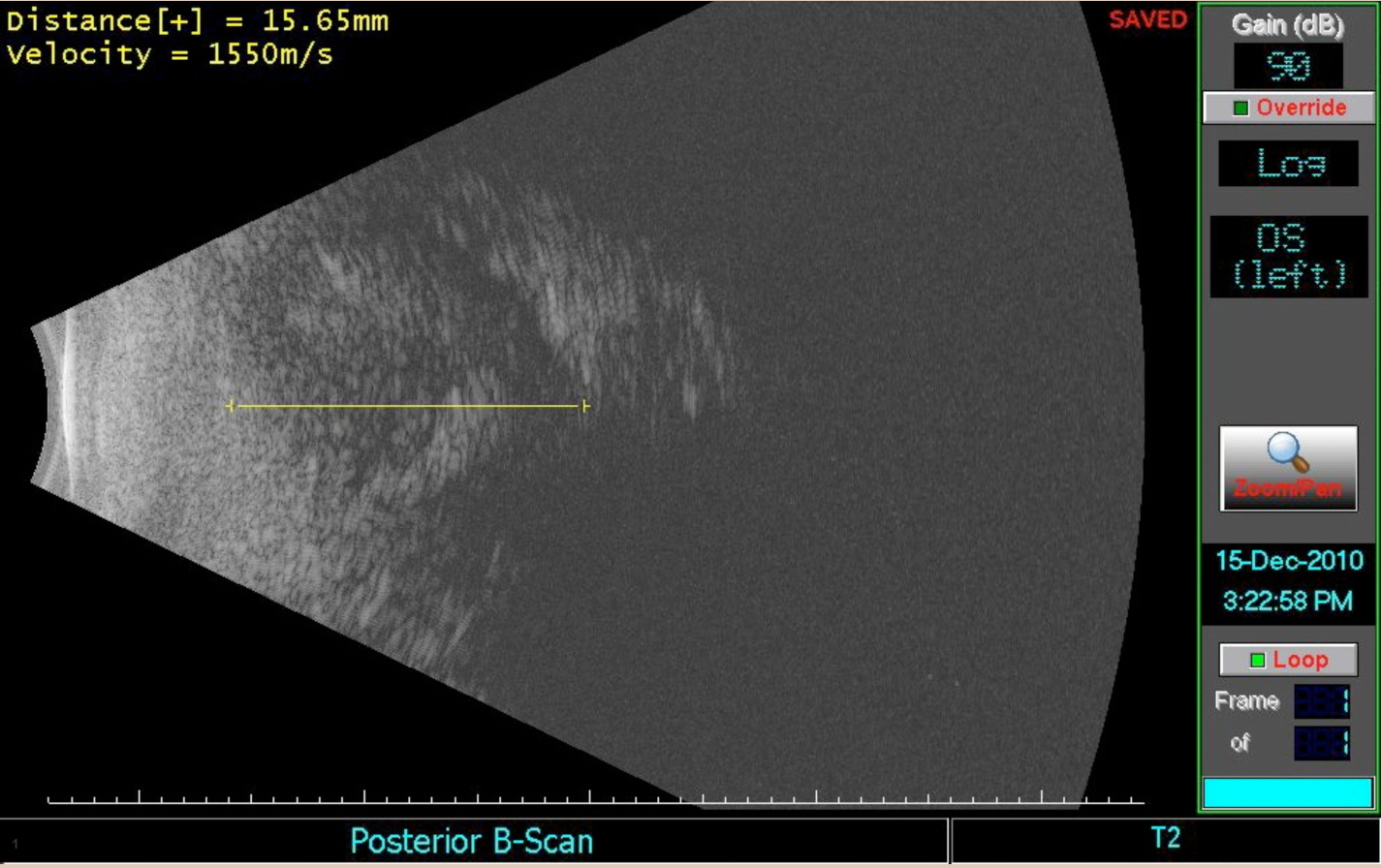
Dilated fundus exam: Cup to disc ratio 0.4 OU, normal macula, vessels, and periphery OU
 |
 |
 |
CLINICAL COURSE
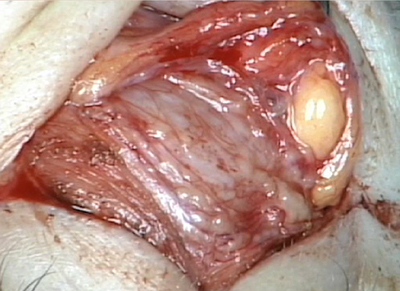
The patient underwent an anterior orbitotomy through an upper lid skin crease incision. {video below} Biopsies of the lacrimal gland were taken.
If video fails to load, use this link: https://vimeo.com/139276839
 |
 |
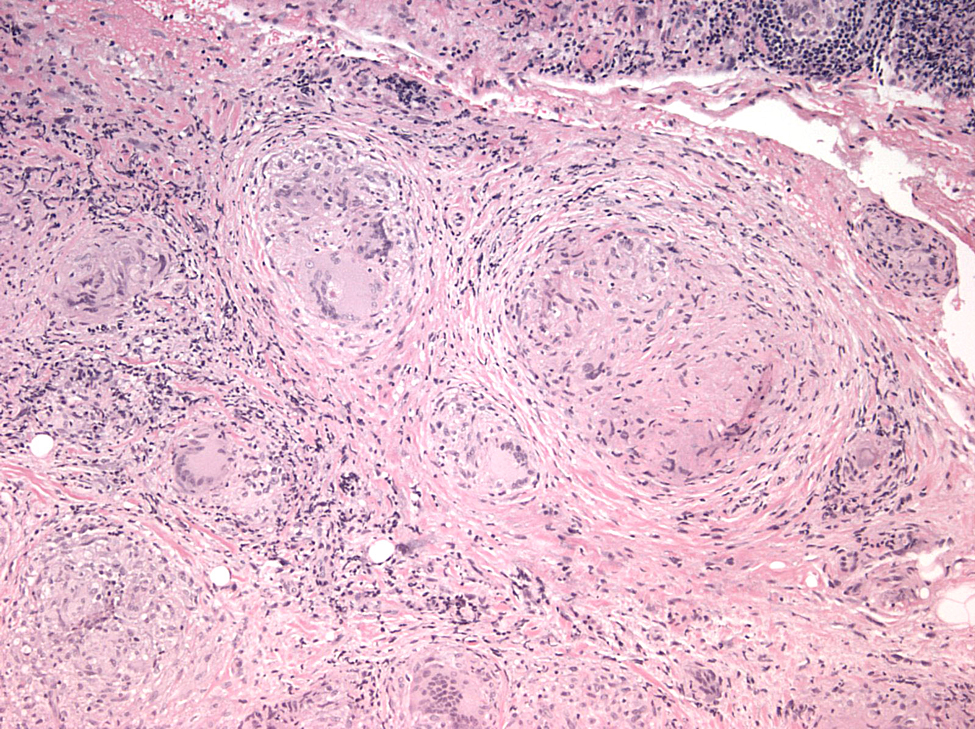 |
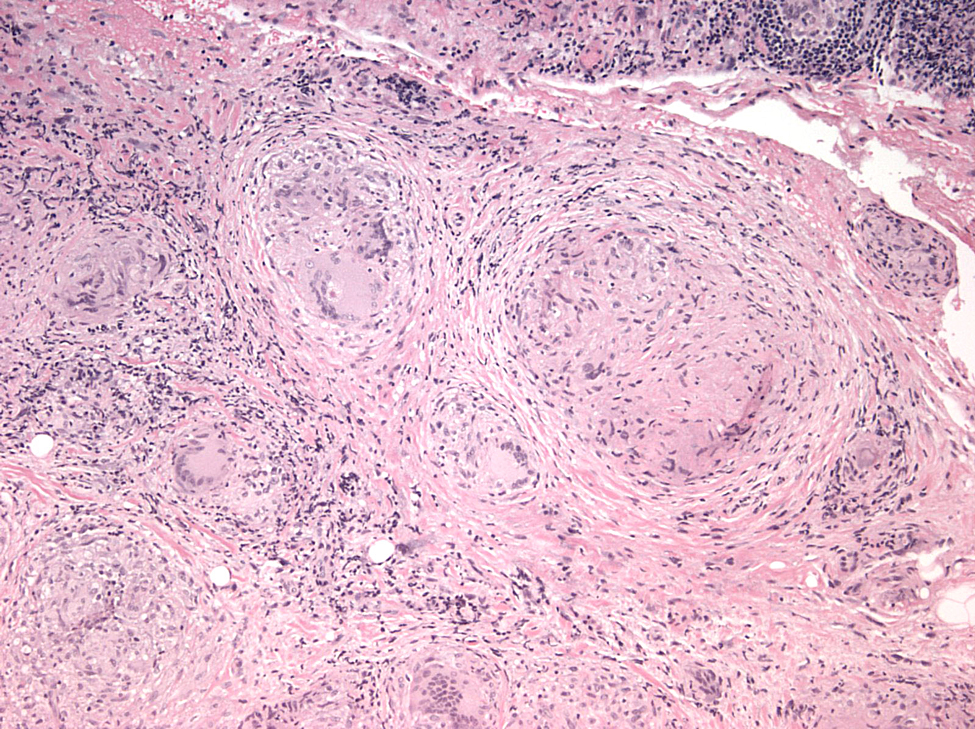
Histopathological analysis of the biopsied tissue revealed diffuse chronic granulomatous inflammation with non-caseating granulomas. The granulomas were characterized by a central core of monocytes, surrounded by macrophages in various states of activation and differentiation (epithelioid histiocytes, large multinucleated giant cells). Scattered lymphocytes were also seen. No normal lacrimal gland tissue was identified. There were no mycobacterial or fungal organisms identified based on negative acid fast and Gomori methanamine silver staining. No foreign material was identified by cross-polarization.
Based upon the histology findings, the diagnosis of sarcoidosis was highly suspected. The following laboratory tests were sent:
| Test | Normal | Results |
|---|---|---|
| ACE | 8-52 U/L |
32 |
| Lysozyme | 9-17 ug/mL |
22 |
| RPR | Non reactive |
Non reactive |
| FTA-Abs | Non reactive |
Non reactive |
| ANCA screen | < 1:40 |
< 1:40 |
A tuberculosis skin test (purified protein derivative) was administered and returned a normal result.
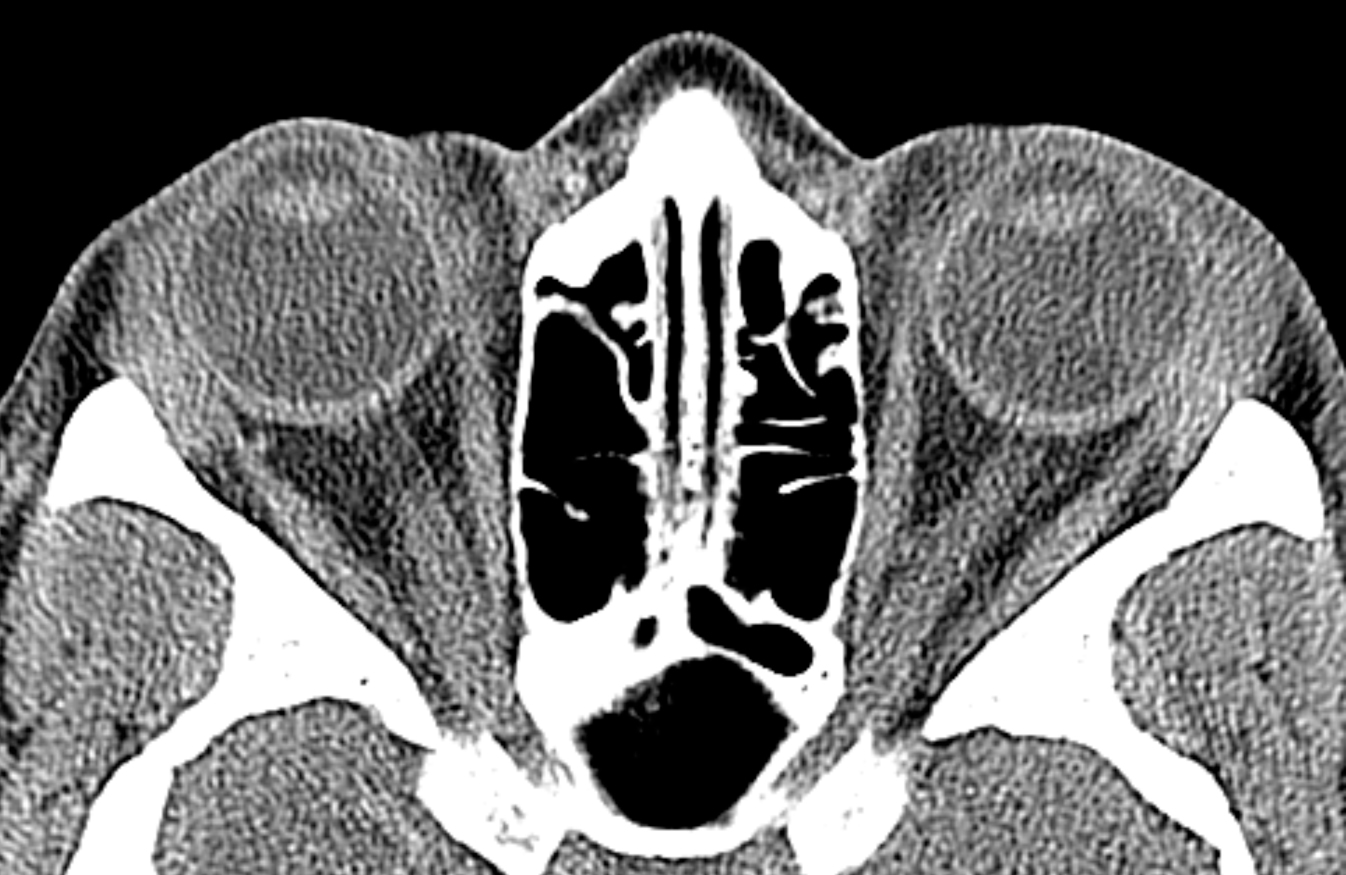
Computed tomography of the chest did not show hilar lymphadenopathy or interstitial infiltrates; however, a small 5mm ground glass nodule was found in the right upper lobe.
She was seen one week post operatively and noted to have persistent, marked inflammation of the left upper eyelid. Although referrals had been made for her to see Rheumatology and Pulmonology before definitive treatment, her appointments were more than a month away. We decided, therefore, to start immunosuppressive treatment with 60mg of oral prednisone, daily.
Two weeks after treatment, her eyelid inflammation had partially improved. We decided to continue oral prednisone until her next visit.
During the interim, she was examined by the pulmonologist. The patient had normal pulmonary function tests, and the lung nodule appeared to be of unclear significance. However, given that she could later develop systemic symptoms, the pulmonology service had planned to follow up in 3 months.
One month after her last visit with our service, the patient had begun experiencing weight gain, insomnia, and irritability from the oral steroids. The eyelid inflammation had not yet resolved completely. Treatment options were presented to the patient, and she elected to receive an intralesional steroid injection of 40mg of triamcinolone and 10mg of dexamethasone.
Sarcoidosis is a multisystem granulomatous disease characterized by T-lymphocyte infiltration, non-caseating granuloma formation, and distortion of the microarchitecture in affected tissues. Despite decades of research on this disorder, the cause of sarcoidosis is still unknown. One possible mechanism for the disorder proposes that individuals with a certain genetic predisposition tend to mount a Th1 response and form granulomas when exposed to an unknown or multiple unknown environmental or infectious agents. Greater concordance for disease in monozygotic twins as compared to dizygotic twins and familial clustering support the case for a genetic predisposition in this disease. The risk of sarcoidosis increases 5-fold if the patient has a first degree relative with the condition (Morganthau 2011). The argument for an environmental trigger is supported by a case-controlled, multicenter, epidemiological study, which showed a modest risk associated with moldy environments and insecticide exposure (Baughman 2001, Iannuzzi 2011, Morganthau 2011). Based on PCR studies within sarcoid tissue, mycobacteria, propionibacteria, HHV 8, and rickettsia have also been proposed as possible causative agents. However, others have tried unsuccessfully to identify bacterial DNA in these same tissues (Jones 2002).
Since there is no absolute diagnostic criterion for this disease, sarcoid remains a diagnosis of exclusion. Often the clinician may be able to say only whether a patient has suspected, presumed, or highly probable sarcoidosis (Weinreb 1984). It is not uncommon for many patients to have a prolonged workup (more than 6 months) prior to being diagnosed with sarcoid (Iannuzzi 2011). The consensus in the literature (Hunninghake 1999, Morgenthau 2010, Iannuzzi 2011) seems to be that one can reasonably diagnose sarcoid if the patient has:
Granulomas are comprised of a central follicle containing epithelioid histiocytes and CD4 type 1 helper T cells surrounded by fibroblasts, B cells, and CD8 T lymphocytes. Co and colleagues describe 4 stages of granuloma development:
Sarcoid, by the most common definitions, is a systemic disease. However ocular and orbital manifestations of sarcoid can precede systemic manifestations by years to decades (Yanardag 2003, Iannuzzi 2011). Additionally, some believe that sarcoid can at times involve only one organ, be self-limiting, and never involve other tissues (Jones 2002, Mavikakis 2007) as evidenced by various case reports and series in the literature, although not all share this perspective (Mombaerts 1996, Raskin 1995, Liu 2003, Prabhakaran 2007). As a result, accurate epidemiological data on this disorder is complicated by the varying ways that different groups have defined “sarcoid”. When sarcoidosis presents acutely as a systemic disease, the diagnosis can be quite straightforward. However, for clinicians who are confronted with either early or limited manifestations of the disease, such as the ophthalmologist, it can be very difficult to make the diagnosis of sarcoidosis. Often the systemic radiologic findings or blood tests may be normal. Some have asked the question: when the disorder affects only the orbital tissues should the condition be called limited sarcoid or solitary granulomatous orbital inflammation (a histologic variant of idiopathic orbital inflammation)? Does it matter to patient care, or is the question merely an academic one?
Given that there is a plethora of literature on systemic manifestations of sarcoidosis, the remainder of this discussion will focus on sarcoidosis affecting the lacrimal gland.
Prabhakaran et al, published a case series of 26 patients (19 female, 7 male) with orbital or adnexal sarcoidosis, with a mean age 57 years (28-83 years) and mean follow up 18.75 months. Eleven of these patients had lacrimal gland disease. The analysis was part of a multicenter retrospective study of patients from orbital centers in Australia, Amsterdam, Texas, West Virginia, and Israel. A patient was diagnosed with sarcoid if there was a biopsy showing non-caseating epithelioid granulomas in the lacrimal gland, orbit, eyelid or lacrimal sac plus 1 or more of the following: hilar lymphadenopathy on chest imaging, non-caseating epithelioid granulomas in another extrapulmonary organ, or an unexplained increase in serum ACE. These authors believe that sarcoidosis is a systemic disease and that sarcoidosis is often confused with isolated orbital granulomatous disease in the ophthalmic literature. They were therefore careful to include only those patients who also had systemic involvement either at the time of diagnosis or later.
In their study, the most common presenting sign in orbital sarcoid is a slowly progressive mass or swelling of the eyelid (65-88.5%) followed by proptosis or globe displacement, discomfort, ptosis, restricted ocular motility, dry eye, diplopia, and decreased vision (Prabhakaran 2007, Mavrikakis 2007).
The lacrimal gland was most common periocular tissue affected (42.3%), followed by other orbital tissues (38.5%), eyelid (11.5%), and lacrimal sac (7.7%).
Of the 11 patients with lacrimal gland involvement who had chest radiographs, 54.5% had hilar lymphadenopathy. A large proportion of these patients had an elevated ACE: 72.5% (81.8% if one includes the elevated CSF ACE found in 1 patient). Serum lysozyme was tested in 8 patients, of whom, only 20% had elevated levels. However, it is unclear whether these 8 had only lacrimal gland disease.
Yanardag and Pamuk described 9 patients with sarcoidosis involving the lacrimal gland in a retrospective case review. Diagnosis of sarcoidosis was based on clinical and radiographic evidence in addition to non-caseating granulomas in at least 1 tissue specimen. Four patients had concurrent systemic abnormalities found on chest x-ray. In 5 of the 9 patients, the chest x-ray was originally normal, so they were diagnosed with isolated lacrimal gland disease. Of these 5 patients, however, all developed other sites of involvement (skin, parotid gland, hilar lymph nodes) over a 36 year period of follow up (Yanardag 2003).
In a retrospective interventional case series of 20 Canadian patients with sarcoidal reactions involving the orbit, 50% of patients were found to have concurrent systemic abnormalities after the ophthalmologic diagnosis was made. Of the 9 patients who were diagnosed with pulmonary disease by chest x-ray or CT, 7 were asymptomatic. Patients were said to have sarcoid of the orbit if the biopsy showed non-caseating granulomas. In contrast to the Prabhakaran study, this study did not exclude patients without systemic disease. The authors found that half of the patients never developed systemic findings (mean of 5 years follow up, range of 6 months to 14 years). An elevated ACE level was seen in only 20% (2/10) of their patients. Both had active systemic disease; however, it is unclear which patients had ACE levels drawn (Mavrikakis 2007).
Below is a table summarizing some of the results pertinent to this discussion:
| Studies with lacrimal gland involvement | Initially normal chest imaging (%) | Elevated serum ACE (%) | Elevated serum lysozyme (%) | Systemic involvement |
|---|---|---|---|---|
| Prabhkaran et al. N = 11 of 26 |
46.5% |
81.8% |
25% (of 8) |
100% |
| Yanardag and Pamuk N = 9 |
55.6% |
NA |
NA |
100% |
| Mavrikakis and Rootman N = 11 of 20 |
50% |
20% (of 10) |
NA |
50% |
Angiotensin I converting enzyme (ACE) is normally found in pulmonary capillary endothelium and in macrophages, particularly epithelioid cells, which are activated macrophages. Given the abundance of epithelioid cells in sarcoid granulomas, it is not surprising that ACE would be elevated in many patients with active systemic sarcoidosis. In addition to ACE, sarcoid granulomas also secrete lysozyme, glucoronidase, collagenase and elastase. The total body mass of epithelioid granulomas is directly related to the level of serum ACE and lysozyme.
Serum ACE. The reported sensitivity of elevated serum ACE levels in sarcoidosis depends upon disease activity in the patients and laboratory assays that were studied. The sensitivity has been reported as low as 29% (Thurton 1979) and 59.0% (Tomita 1999) and as high as 60-90% in active disease (Weinreb 1984), but is 95% specific (in patients with uveitis) (Baarsma 1987). ACE can also be elevated in primary biliary cirrhosis, Gaucher's disease, leprosy, histoplasmosis, hyperthyroidism, diabetes mellitus and histiocytic medullary reticulosis. It is also elevated in children when compared to adults. Interestingly, immunoflouorescent examination of ACE in sarcoid granulomas might be more specific. In 38 of 39 granulomas from sarcoid patient, ACE was detected, but it was not detected in the nonsarcoid granulomas (Pertschuk 1981).
Serum lysozyme. The sensitivity of an elevated serum lysozyme for predicting sarcoidosis is about 69% (Turton 1979) to 79.1% and increases with the number of organs involved (p < 0.01) (Tomita 1999). Tomita et al studied 110 Japanese patients who had sarcoidosis diagnosed by epithelioid granulomas in the biopsy specimens from the lung, skin or lymph nodes. The majority of patients had at least hilar lymphadenopathy on chest radiography (89%). The control subjects had other granulomatous disease such as hypersensitivity pneumonitis, tuberculosis and aspergillosis. In this study, the mean serum lysozyme among the sarcoidosis subjects at the initial visit was 15.7 ± 7.0 µg/ml and 18.4 ± 7.7 µg/ml at the maximum. Patients that had more extensive pulmonary involvement had higher lysozyme levels. Its level usually correlates with that of serum ACE but is more sensitive and less specific when compared in the same study (Turton 1979, Tomita 1999). Others have noted that the lysozyme can be elevated when the ACE is not (Romer 1982), and in the Tomita et al study, the sensitivity of lysozyme among patients without an elevated ACE was 72.1% (31 of 43 patients). One proposed reason for the discrepancy between ACE and lysozyme levels may be due to the difference in molecular weight and rates of diffusion into the blood stream—ACE is about 10-fold larger than lysozyme (Prior 1990). Lysozyme can also be elevated in leprosy, tuberculosis, pernicious anemia, osteoarthritis, monocytic and myelomonocytic leukemias, histiocytic medullary reticulosis, and renal insufficiency. Specificity of the test is from 60% (Tomita 1999) to 76% in patients with uveitis (Baarsma 1987). Lysozyme often decreases in patients who are treated with immunosuppression and can therefore be used to follow disease activity.
Serum and urine calcium. Sarcoid patients have increased production and activity of vitamin D, which leads to increased absorption of calcium from the intestines. Approximately 2-10% of sarcoid patients have hypercalcemia. Hypercalciuria is twice as common as hypercalcemia. The test has limited diagnostic value, however, because it is not specific for sarcoid (Weinreb 1984).
In our patient, we decided to order a serum ACE and lysozyme. Her ACE level returned normal, but her lysozyme appeared to be only slightly elevated at 22 µg/ml. However, when compared to the levels obtained in the Tomita et al study, her lab result is well within the range of those patients who had an elevated lysozyme in the setting of known sarcoidosis. This result may be consistent with early, limited disease at the time of testing.
Chest X-Ray. Chest x-ray findings are staged based on the following abnormalities: 1) stage 0 normal, 2) stage 1 hilar lymphadenopathy alone, 3) stage 2 hilar lymphadenopathy with parenchymal involvement, 4) stage 3 parenchymal involvement without adenopathy, and 5) stage 4 fulminant fibrosis.
High resolution chest CT: HRCT is sensitive for mediastinal lymph node and parenchymal infiltration and correlates well with positive lung biopsy (Kaiser 2002). Characteristic findings on chest CT include small nodules and irregular linear densities along the bronchovascular bundles (Muller 1989). HRCT may have the same diagnostic accuracy as a lung biopsy (Takahashi 2001).
Our patient did have an abnormality on her chest CT; however, it was not a commonly found lesion in patients with pulmonary sarcoidosis.
Pulmonary function tests. PFTs are a sensitive test of granulomatous infiltration in the lungs and may be abnormal even before finding on chest X-ray. One of the earliest signs is reduced diffusion capacity (Weinreb 1984).
Gallium scan. Gallium citrate correlates with T lymphocyte infiltration in inflammatory lung disease. It is more sensitive that chest x-ray. However, it can be positive in Sjogren's syndrome, lymphoma, carcinoma, tuberculosis and silicosis. Up to 90% of patients with active sarcoid may demonstrate increased lacrimal gland uptake (Weinreb 1984).
F-18 Fluorodeoxyglucose (FDG) PET scanning can be valuable in those patients with sarcoidosis who have no readily identifiable tissue to biopsy. In the setting of cardiac sarcoidosis, FDG PET scanning was 82-100% sensitive and 82-91% specific (Morganthau 2011).
Conjunctiva. When conjunctival biopsies are performed without a defined lesion, 7-55% are positive (Bonfioli 2005) but most report a yield of less than 10% (Weinreb 1987). If there are visible lesions, 85% of the biopsies are positive (Bonfioli 2005). The general consensus is to biopsy visible conjunctival lesions or to biopsy when the suspicion for sarcoidosis is high, but no lesion has been identified.
Minor salivatory gland. Biopsy of the minor salivatory glands in the oral mucosa have a 58% yield in patients with other clinical symptoms or signs compatible with sarcoid (Weinreb 1987).
Other common locations to biopsy include the skin and pulmonary tissue via transbronchial biopsy of hilar lymph node or open lung biopsy.
It is important to note that granuloma formation is not absolutely specific for sarcoidosis. Unless other clinical symptoms and signs support the diagnosis, a biopsy may not be enough to secure the diagnosis.
Corticosteroids remain the most accepted treatment for sarcoidosis. For systemic symptoms, the recommended dosage is 30-40mg/day of oral prednisone with a biweekly taper until 10-20mg/day, at which point the dose is maintained for months. Treatment usually lasts for a total of 6-9 months (Iannuzzi 2011). In the ophthalmological literature, doses of up to 60-80mg of oral prednisone (1mg/kg) tapered over 3 months or periocular injections of 1ml of 40mg/ml triamcinolone have been used (Prabhakaran 2007) for localized disease.
Methotrexate has been shown in a randomized double-blind placebo-controlled trial with 24 patients with pulmonary sarcoidosis to decrease the concomitant need for prednisone at 12 months (Baughman 2000). So far, cyclosporine and the tumor necrosis factor (TNF) inhibitor etanercept have not shown clinical benefit in the treatment of pulmonary sarcoidosis; however infliximab, another TNF inhibitor, has shown a small but statistically significant benefit (Iannuzzi 2011). Infliximab has also shown some benefit in refractory cases of ocular sarcoidosis (Margolis 2007). Interestingly, statins, which are known to regulate certain transcription factors in inflammatory responses, are being tested in a double-blind randomized controlled trial at the NIH to see if they have steroid sparing effects in patient with pulmonary sarcoid.
If the eye or orbit is involved, sarcoidosis is usually treated. However, it is important to realize that some patients with systemic sarcoid may not need treatment. Pulmonologists will often treat only if troublesome symptoms such as intractable coughing, dyspnea on exertion or progressive pulmonary disease are present.
Perhaps the most important reason for correctly identifying an orbital lesion as sarcoid rather than idiopathic orbital inflammation is that those with the former should have long term follow up by a pulmonologist or rheumatologist because systemic disease can develop months to years after orbital disease and because steroid responsive cases are at increased risk for relapses (Prabhakaran 2007, Yanerdag 2003).
Half of all patients can achieve remission within 3 years and 2/3 within 10 years. However, 1/3 of patients have persistent disease leading to significant impairment. Less than 5% die, usually from cardiac or respiratory failure or neurological disease (Iannuzzi 2011). Pulmonary fibrosis is the most common cause of death in this disease.
Because sarcoidosis is a diagnosis of exclusion, when the lacrimal gland is the only involved organ, one must rule out infection (tuberculosis, atypical mycobacteria, syphilis, fungal), neoplasm, and other known inflammatory disorders (ANCA associated inflammation, lupus).
Lacrimal gland involvement can be the initial sign in sarcoidosis. As ophthalmologists, we are often catching the disease at an early stage when ACE levels and chest radiography may be normal. Studies that have followed patients with orbital involvement for many years have found systemic symptoms during subsequent examinations. Therefore, it is important to arrange long-term follow up for these patients.
There is controversy between whether lacrimal gland involvement characterized by sarcoidal reaction in the absence of systemic signs is actually a form of idiopathic orbital inflammation. The distinction can be difficult to resolve initially because both are idiopathic and diagnoses of exclusion and because early manifestations of sarcoidosis may involve the lacrimal gland only. Nevertheless, the treatment for both disorders is corticosteroid therapy. The controversy will likely continue until a specific etiology for sarcoidosis is discovered.
EPIDEMIOLOGY:
|
SIGNS:
|
SYMPTOMS:
|
TREATMENT:
|
Barsma GS, La Hey E, Glasius E. The predictive value of serum angiotensin converting enzyme and lysozyme for the diagnosis of ocular sarcoidosis. Am J Ophthalmol 1987; 104:211-217.
Baughman RP, Winget DB, Lower EE. Methotrexate is steroid sparing in acute sarcoidosis: results of a double blind, randomized trial. Sarcoisosis Vasc Diffuse Lung Dis. 2000;17(1):60-66.
Bonfioli AA and Orefice F. Sarcoidosis. Semin Ophthalmol 2005; 20:177-182.
Bresnitz EA and Strom EB. Epidemiology of sarcoidosis. Epidemiol Rev 1983;5:124-56.
Collison JMT, Miller NR, Green WK. Involvement of orbital tissues by sarcoid. Am J Ophthalmol 1986; 122:302-207.
Hunninghake GW et al. ATS/ERS/WASOG statement on sarcoidosis. American Thoracic Society/European Respiratory Society/World Association of Sarcoidosis and other Granulomatous Disorders. Sarcoidosis Vasc Diffuse Lung Dis. 1999; 16(2):149-173.
Iannuzzi MC and Fontana JR. Sarcoidosis: clinical presentation, immunopathogenesis, and therapeutics. JAMA. 2011; 305(4): 391-399
Jones NP. Sarcoidosis. Current Opinion in Ophthalmology 2002; 13:393-396.
Kaiser PK, et al. Chest computerized tomography in the evalutation of uveitis in elderly women. Am J Ophthalmol 2001; 133:499-505.
Liu CH et al. Idiopathic granulomatous orbital inflammation. Chang Gung Med J 2003; 26:847-50.
Margolis R and Lowder CY. Sarcoidosis. Curr Opin Ophthalmol 2007; 18:470-475.
Mavrikakis I and Rootman J. Diverse clinical presentations of orbital sarcoid. Am J Ophthalmol 2007; 144:769-775.
Mombaerts I, et al. Idiopathic granulomatous orbital inflammation. Ophthalmology 1996; 103(12):2135-2141.
Morgenthau AS and Iannuzzi MC. Recent Advances in Sarcoidosis. Chest 2011; 139:174-182.
Muller NL, Kullnig P, Miller RR. The CT findings of pulmonary sarcoidosis: analysis of 25 patients. American Journal of Roentgenology, 1989; 152(6):1179-1182.
Pertschuk LP, Silverstein E, Freidland J. Immunohistological diagnosis of sarcoidosis. Am J Clin Path 1981; 75:350-354
Prabhakaran VC, et al. Orbital and Adnexal Sarcoidosis. Arch Ophthalmology 2007; 125(12):1657-1662.
Raskin EM, et al. Granulomatous idiopathic orbital inflammation. Ophthalmic Plast Reconstr Surg 1995; 11:131-5.
Rothova A. Ocular involvement in sarcoidosis. Br J Ophthalmol 2000; 84:110-116.
Takahashi et al. Significance of lymphocystosis in bronchoalveolar lavage in suspected ocular sarcoidosis. Eur Respir J 2001; 18:515-521.
Tomita H, et al. Serum lysozyme levels and clinical features of sarcoidosis. Lung 1999; 177:161-167.
Turton CWG, et al. Value of measuring serum angiotensin I converting enzyme and serum lysozyme in the management of sarcoidosis. Thorax 1979; 34:57-62.
Weinreb RN and Tessler H. Laboratory diagnosis of ophthalmic sarcoidosis. Surv Ophthalmol 1984; 28:653-664.
Yanardag H and Pamuk ON. Lacrimal gland involvement in sarcoidosis. Swiss Med Wkly 2003; 133:388-391.
Tsui JY, Allen R. Sarcoidosis Affecting the Lacrimal Gland. EyeRounds.org. July 14, 2011. Available from: https://eyerounds.org/cases/135-sarcoidosis-lacrimal-gland.htm

Ophthalmic Atlas Images by EyeRounds.org, The University of Iowa are licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License.

Address
University of IowaLegal
Related Links
