
A case of an 81-year-old woman with limited eye movements
Chief complaint: Abnormal eye movements
The patient was accompanied by her husband, who provided a substantial portion of the history.
An 81-year-old woman presented to the Neuro-Ophthalmology clinic at the University of Iowa Hospitals and Clinics with increasing difficulty moving her eyes. Her difficulty was first observed by her husband 1-2 years ago, and had steadily worsened since onset. Looking upward and downward were most impaired, but all directions were affected to some extent. The patient’s husband was concerned that her inability to look downward caused her to frequently trip over bumps in the ground. These stumbles caused the patient, who already walked with a wide, awkward gait, to fall over backwards. Despite her limited eye movements, the patient had not noted diplopia and denied vision changes.
The patient had developed difficulties with speaking over the same time period as her gaze difficulties, and she was diagnosed with Progressive Non-Fluent Aphasia by a local neurologist. Her verbal responses had deteriorated to 1-2 word phrases by the time she presented to Neuro-Ophthalmology clinic, and she had developed a several second delay in her responses. Her ability to orally recite written material had degenerated to naming individual letters, but she retained an ability to understand verbal instructions and books read aloud. The patient also had mild difficulties with memory but her husband had not noticed any loss of social skills or changes in personality.
Past Medical History: Progressive Non-Fluent Aphasia as noted above. Additional history was significant for hypertension, hyperlipidemia, anxiety, and restless leg syndrome.
Medications: alprazolam, aspirin, atorvastatin, hydrochlorothiazide, omeprazole, oxycodone, pregabalin
Family History: non-contributory, including no history of eye or neurologic problems
Social History: no alcohol or tobacco use, married homemaker for the last 60 years
Visual Fields: could not be obtained due to difficulties with patient communication
Pupils: 3 to 2 with minimal reaction to light, both eyes (OU)
 |
 |
 |
 |
Dilated Fundus Exam: Thinning of optic disc inferiorly OD, superiorly and inferiorly OS; cup-to-dic ratio 0.5 OD, 0.8 OS. The maculae, vessel, and periphery were normal.
Based on time course, symptoms, and signs, the patient was diagnosed with Progressive Supranuclear Palsy. The diagnosis was explained to the patient, and she and her husband were counseled on the prognosis of the condition, expected time course for symptom progression, and options for future care. The patient was referred to in-house neurology, who confirmed the findings of Progessive Supranuclear Palsy and Progessive Non-Fluent Aphasia. Neurology also conducted an extensive evaluation of the patient’s right arm weakness and noted features of Corticobasal Degeneration, specifically right hand apraxia, exertion tremor, reduced stereognosis, and alien limb phenomenon. The patient’s final diagnosis was of a mixed clinical tauopathy with elements of Progressive Supranuclear Palsy, Progressive Non-Fluent Aphasia, and Corticobasal Degeneration. Follow-up clinic visits were arranged with both neurology and neuro-ophthalmology, and MRI brain imaging was ordered at time of follow-up. The patient was also advised to use artificial tears for her dry eyes and to use separate pairs of near and far vision glasses in place of bifocals.
Progressive Supranuclear Palsy (PSP) is an uncommon neurodegenerative condition, first described by Steele, Richardson, and Olszewski in 1964 (1). The classical findings in PSP include vertical gaze palsy, postural instability, axial rigidity and behavioral changes. PSP is often considered an atypical parkinsonian disorder, along with multiple system atrophy and corticobasal degeneration. Estimates of prevalence fall between 6-7 per 100,000 (2). Onset of symptoms occurs after age 40, with peak onset at age 63 (2). It is unclear whether men or women are more commonly affected (3). Though genetic factors may confer susceptibility to the disease, PSP cases are usually sporadic and not inherited (4).
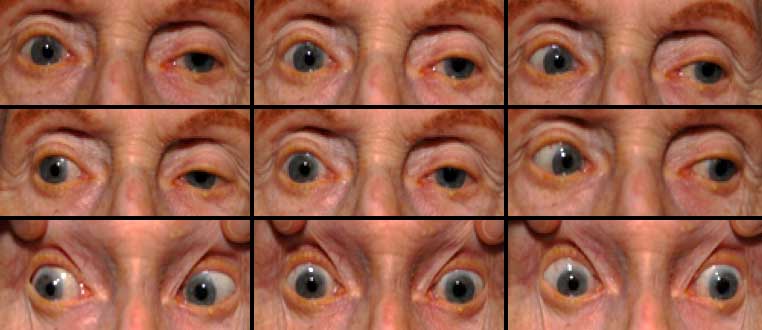
Clinical findings in PSP may be slight at first presentation but become more pronounced as the disease progresses. The eye movements affected by PSP require supranuclear input (as the name PSP suggests) and include voluntary saccades, which are usually affected first, smooth pursuit, and vergence (2). Both the range and velocity of these movements are decreased, and movements in the vertical direction are more affected initially. Eye-movements requiring only lower motor neurons, such as the vestibulo-ocular (Doll’s eye) reflex, are spared (4). Patients may also exhibit square wave jerks, which are brief, horizontal, saccadic intrusions into fixation. A combination of decreased blink frequency and lid retraction often gives these patients a surprised or startled appearance and can lead to dry eyes. Additional ocular findings include photophobia, blepharospasm, and eyelid opening apraxia (2).
Though patients are most likely to present with eye findings in the setting of an ophthalmology practice, key neurologic findings are also apparent on exam and should be thoroughly assessed. Gait disturbance is common, and patients can present with shuffling or lurching gait early in the course of the disease. Coupled with the inability to look downward, this gait apraxia commonly leads to falls in PSP patients (5). The falls are typically backwards, and little effort is made by the patient to break his or her fall, giving the patient a “toppling bowling pin” appearance. Rigidity of the neck and other axial muscles is common and can be detected during examination of the vestibulo-ocular reflex. Patients may also experiences changes in behavior and personality, most often apathy, disinhibition, or anxiety (3). Finally, PSP patients may exhibit frontal release signs, including the grasp reflex and the applause sign, which is the inability to clap only three times on command (2).
Diagnosis of PSP is presumptive and based on clinical findings, as a definitive diagnosis requires histologic examination of brain tissue. Clinical criteria are:
 |
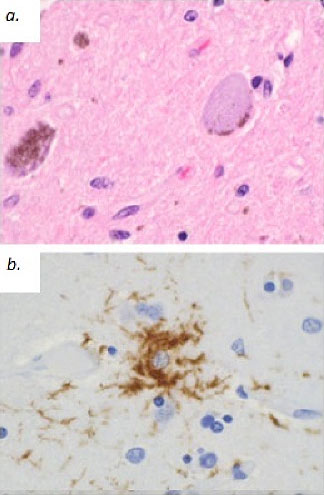 |
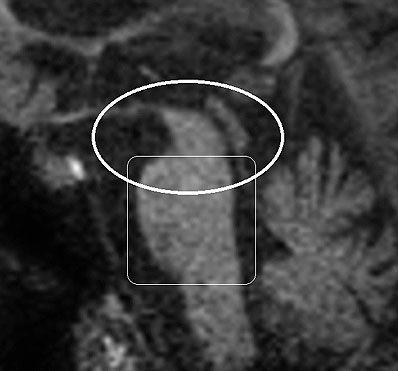
Patients must meet criteria A, as well as either B or C (2). Poor response of symptoms to levodopa therapy may aid in diagnosis. Diagnosis of PSP should not be applied to patients whose symptoms better fit other neurodegenerative disorders, such as corticobasal degeneration (CBD), multiple system atrophy (MSA), Alzheimer’s disease, Parkinson’s disease, or Lewy body dementia. Post-mortem examination commonly shows marked midbrain atrophy with relative sparing of surrounding structures (4). This atrophy may also be apparent on MRI imaging in the late stages of the disease and is described as the “hummingbird sign (figure 3) (6).” Histologic examination shows deposits of the 4R isoform of tau protein throughout the midbrain and to a lesser extent in the basal ganglia, diencephalon, and brainstem. Tau inclusions typically appear as globose neuro-fibrillary tangles in neurons, “tufted” astrocytes, and coiled bodies in oligodendrocytes and can be appreciated with silver or immunohistochemical staining (figure 4) (3). PSP is one of several neurodegenerative diseases, including CBD, MSA, and frontotemporal dementia (FTD), in which tau has been implicated in the pathogenic process. Overlap between these diseases is not uncommon, as in the above case where an overlap between PSP and a type of FTD called Progressive Non-Fluent Aphasia (PNFA) is demonstrated (3). In such cases, patients usually present with word finding difficulties and subsequently develop the classic PSP findings (5). Whether these syndromes represent different faces of a common disease process has not yet been fully demonstrated, but several studies suggest the clinical picture and histological findings may overlap as well (7, 8, 9). To the Authors’ knowledge, no association between tauopathies and the development of open angle glaucoma has been conclusively established. However, a recent study demonstrated increased levels of an abnormal form of tau in retinal and optic nerve tissue samples in patients with increased intra-ocular pressure (10).
At present, PSP has no known cure and no therapies are known to slow its progress. Pharmacologic agents are of little help in alleviating symptoms; in particular poor response to levodopa is suggestive of the diagnosis. Supportive care is of the greatest help to patients. Patients should avoid using bifocals and instead use one set of glasses for distance vision and another for near vision activities because of their inability to look down. Prisms may be placed in the near vision glasses to correct for convergence insufficiency when appropriate. Meals and other items/activities of interest should be placed at eye level to aid visualization. Glasses with vertical prism may be used to shift the patient’s field of view inferiorly, if desired. Artificial tears can be helpful in patients with decreased blink, while eyelid crutches can assist patients with eyelid opening apraxia. Botulinum injections can be used in patients with blepharospasm (2). Walking aids and physical therapy may help the patient to manage postural instability, and occupational therapy can help the patient carry out activities of daily living. Speech therapy should be considered in patients with swallowing difficulties. Patients are usually care-dependent within 3-4 years of initial presentation (3). Median survival is estimated at 6 years from symptom onset (2).
Epidemiology
|
Signs/Symptoms
|
Diagnosis
|
Treatment and Course
|
Null RC, Kwon YH, Thurtell MJ. Progressive Supranuclear Palsy in a Patient with Progressive Nonfluent Aphasia. EyeRounds.org. August 29, 2012; Available from: https://eyerounds.org/cases/155-progressive-supranuclear-palsy.htm

Ophthalmic Atlas Images by EyeRounds.org, The University of Iowa are licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License.

Address
University of IowaLegal
Related Links
