
Chief Complaint: Light sensitivity during outdoor activity
History of Present Illness: A previously healthy 12-year-old boy noticed a 6 to 12 month history of progressive photophobia during outdoor activities. Despite increasing use of sunglasses, his symptoms had worsened. More recently, he had become symptomatic indoors, particularly while watching television. In addition, he had intermittent redness and mild foreign body sensation in both eyes (OU) that was only mildly alleviated with the intermittent use of topical lubrication.
Past Ocular History: No prior history of ocular disorders or therapeutic interventions. He does not wear glasses or contacts.
Medical History: None
Review of Systems: Negative
Medications: Artificial tears OU as needed
Family History: No known ocular disease in either parent or the patient's 16-year-old sister. The mother emigrated from France as a child and the father was adopted.
Social History: Noncontributory.
 |
 |
 |
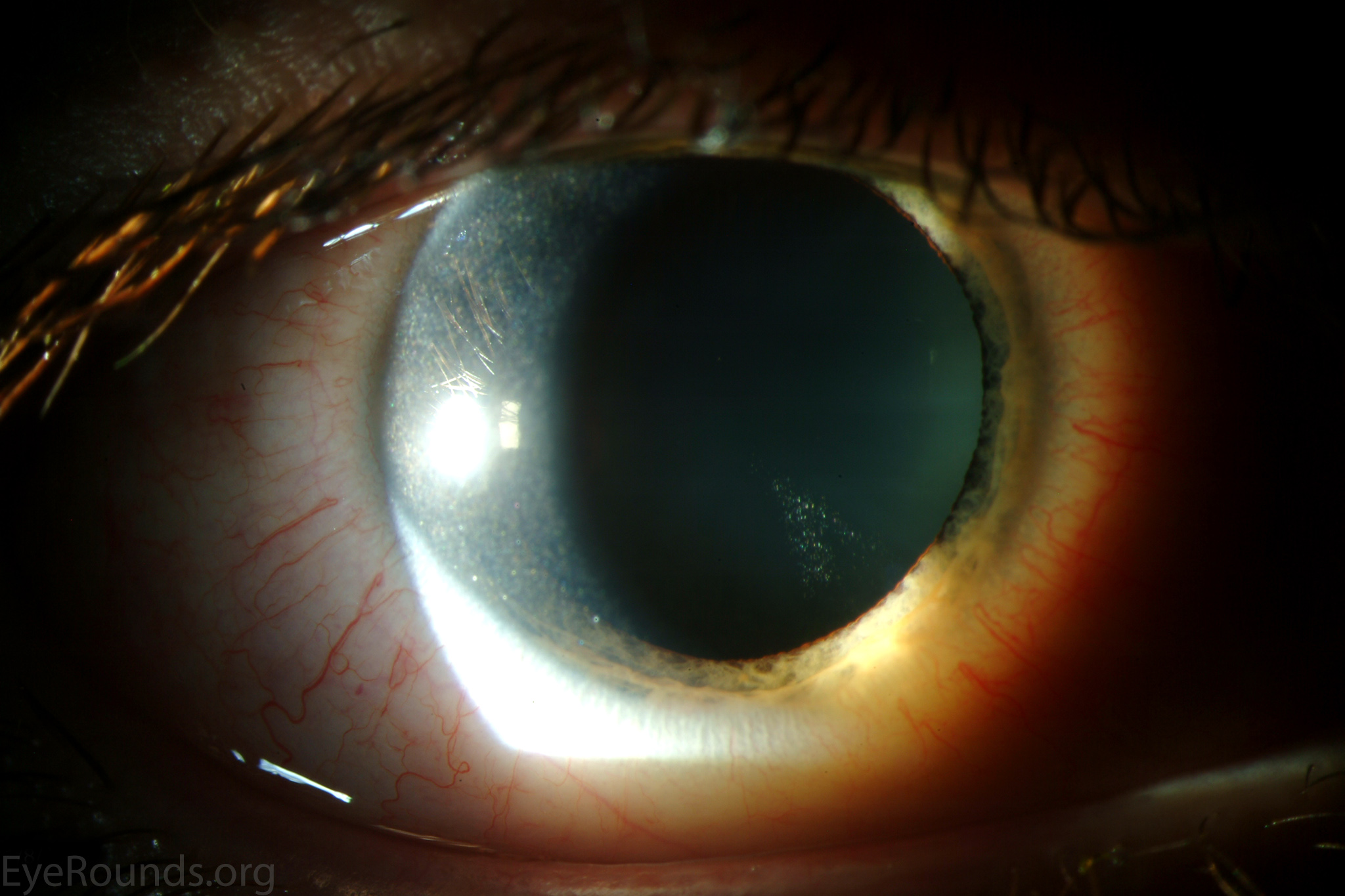 |
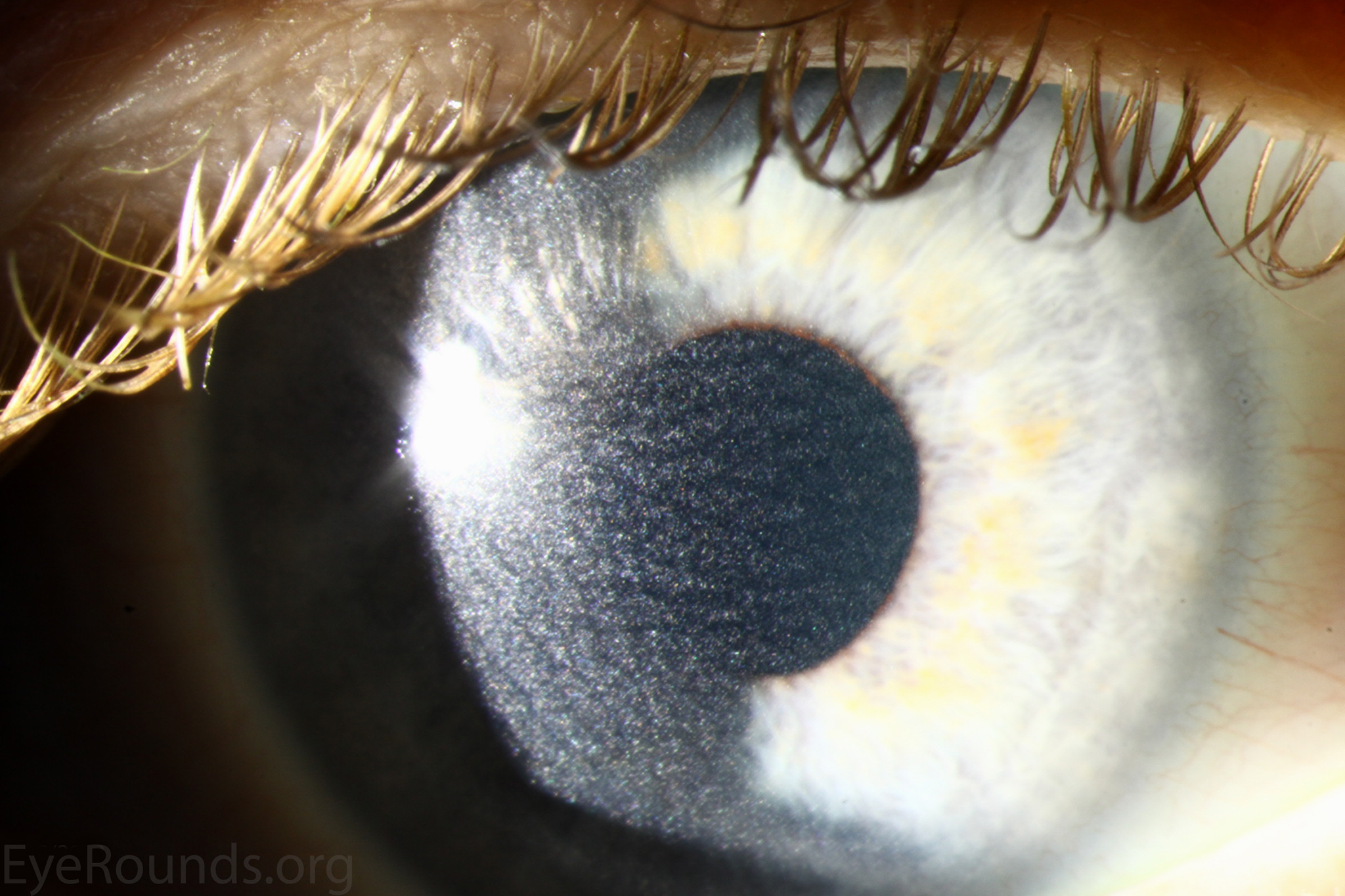
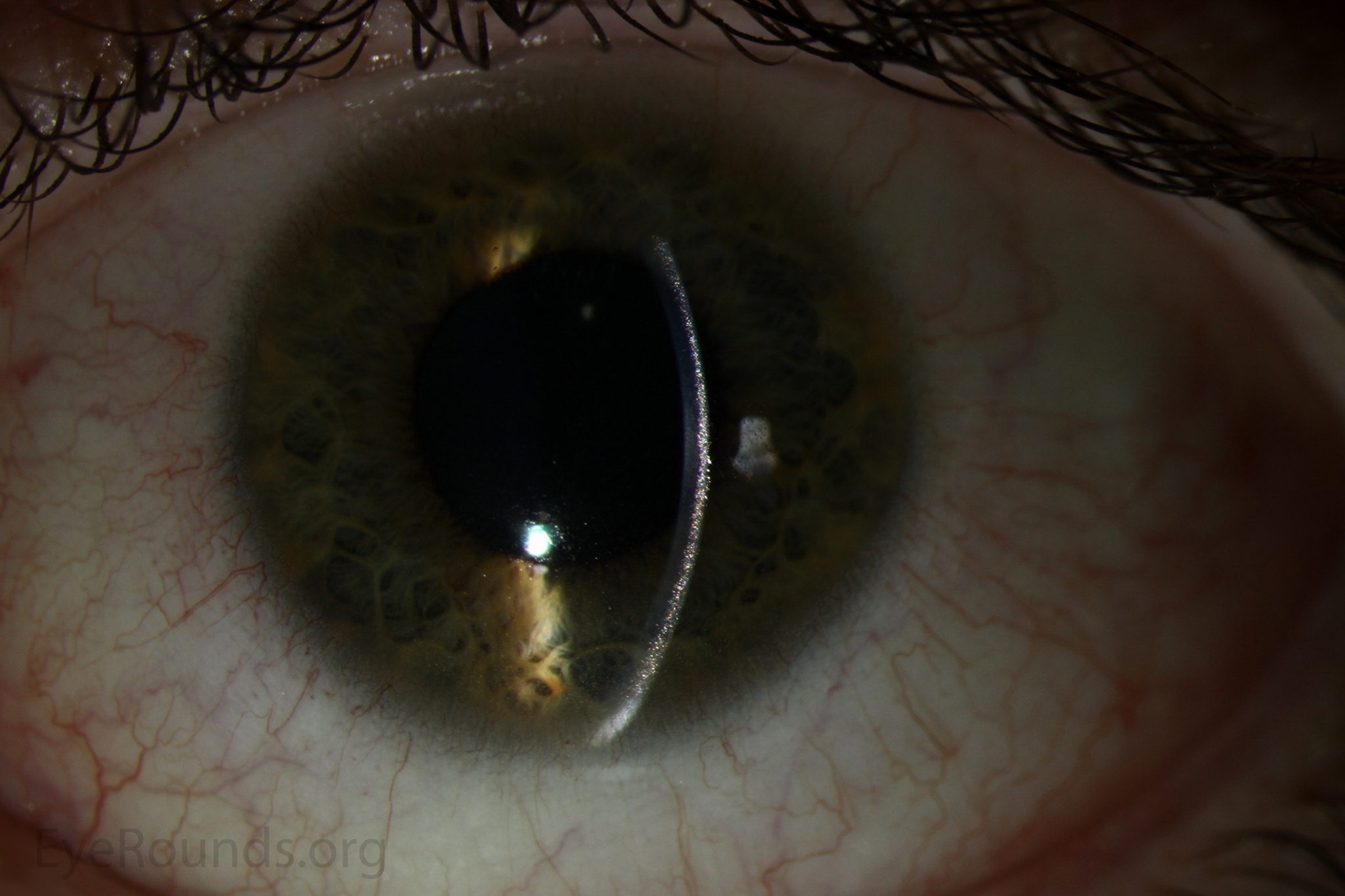
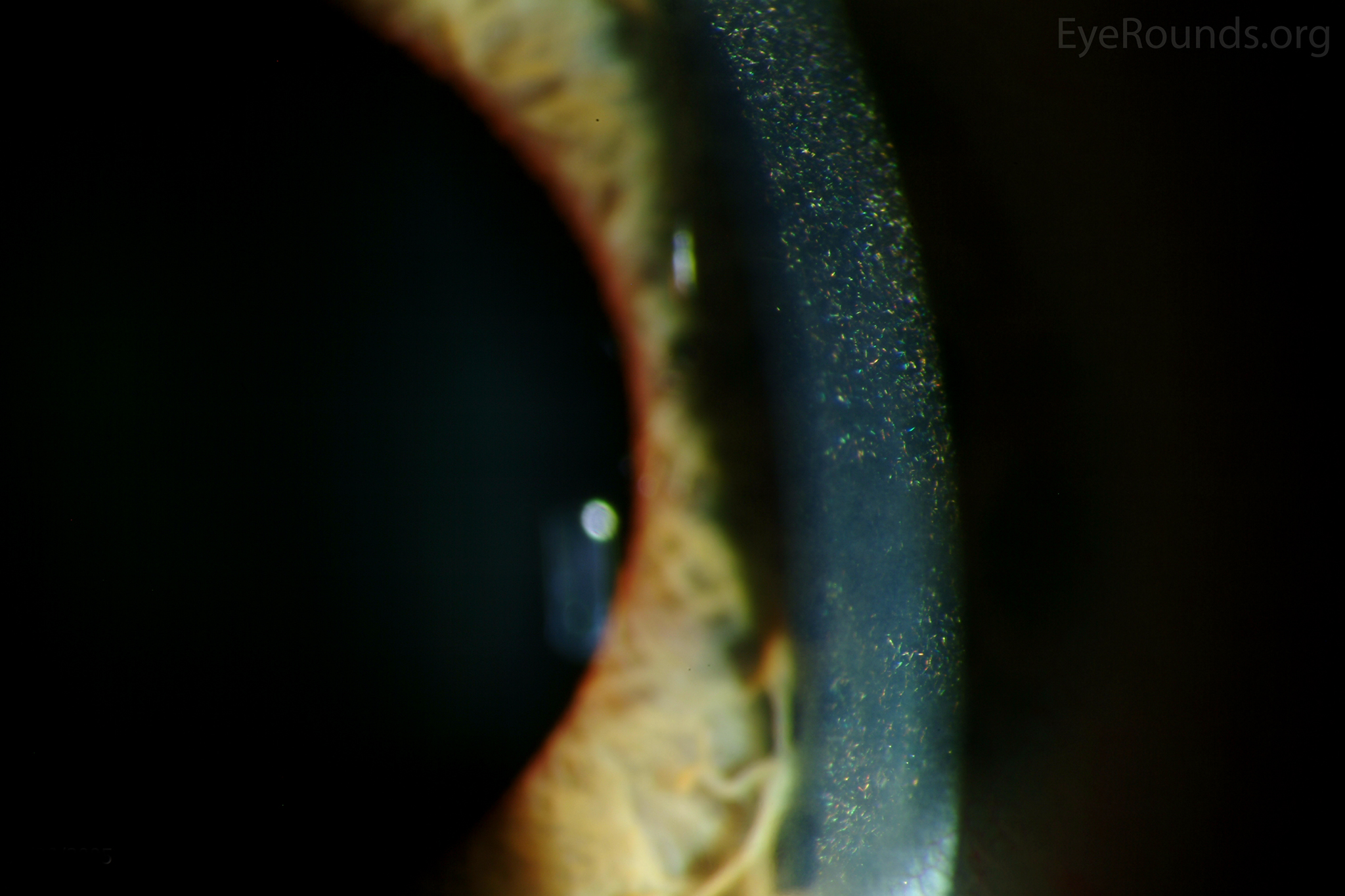
Figure 1. Punctate, needle-shaped crystals diffusely present throughout the corneal surface. They are mostly localized to the epithelium and anterior stroma as evident with the slit beam. Crystals initially appear in the corneal anterior periphery and move posteriorly and centrally as they accumulate (1). The crystals may also appear on the endothelium and throughout the anterior chamber such as on the iris or anterior lens capsule as demonstrated here.
The presence of corneal crystals, in conjunction with increased levels of leukocyte cystine, leads to the diagnosis of cystinosis. The onset of ocular symptoms, in the absence of systemic signs and symptoms, is consistent with a diagnosis of intermediate (juvenile or late-onset) nephropathic cystinosis.
The patient was started on cysteamine HCl 0.44% (Cystaran®) ophthalmic solution every hour while awake and he was referred to a pediatric nephrologist who prescribed cysteamine bitartrate (Cystagon®) 450 mg orally, 4 times daily. The leukocyte cystine measurements were reduced to < 1.0 nmol half-cystine/mg protein 5 hours after taking medication.
Over the next 20 years, the visual acuity remained 20/20 in both eyes, with complete resolution of all subjective symptoms. He eventually developed renal involvement and required renal transplantation at age 25 due to end-stage renal disease. He did not develop any other systemic manifestations of the disorder.
Cystinosis is a metabolic disorder resulting from autosomal-recessive inherited genetic defects in the CTNS gene (2). Cystinosis has been attributed to 112 identified mutations, including missense, nonsense and splice mutations, deletions and insertions (2,3). In patients affected by cystinosis, transport of the amino acid cystine from inside lysosomes is impeded due to improper functioning of protein transporter coded for by the CTNS. Following routine protein degradation inside the lysosome, cystine accumulates instead of being transported outside of the lysosome and recycled. Cystine is poorly soluble and form crystals with accumulation. This leads to cell damage, cell death and eventual organ damage (4-8).
Three distinct forms of cystinosis exist. All three forms can result in anterior segment crystal deposition, but only the nephropathic forms affect the posterior segment (9).
1. Nephropathic cystinosis, also known as infantile, classic and early-onset cystinosis, is the most severe form of the disease. It typically present in the first 6-12 months of life with renal involvement including Fanconi syndrome (disease of the proximal renal tubules resulting in glycosuria, metabolic acidosis, phosphaturia, and aminoaciduria) and renal loss of salt and water (1,10). All cases require renal transplantation in childhood (10). Growth failure is also a common early finding. The eyes are often affected early in the disease course with cystine crystals depositing in the cornea causing photophobia, blepharospasm and occasionally corneal erosions. Crystals also deposit in the conjunctiva, iris, trabecular meshwork, anterior capsule, optic nerve and retina. Though irritating, the corneal crystal deposits do not affect visual acuity (1,10). Retina abnormalities occur in some patients as early as infancy and include retinal pigment epithelium hypopigmentation, appearing as a "salt and pepper" type fundus which may result in an abnormal visual field (11). Other organs affected include the thyroid, pancreas, muscle and central nervous system (10).
2. Intermediate nephropathic cystinosis, also known as juvenile or late-onset cystinosis, is less common than the infantile form and includes the same spectrum of disease but with later onset and less severity, particularly with respect to the systemic manifestations. Nonetheless, end-stage renal disease, such as that which occurred in our patient, may occur, requiring transplantation in early adulthood (2).
3. Non-nephropathic cystinosis, also known as adult or ocular cystinosis results in isolated ocular signs and symptoms and do not experience the effects on other organs (2).
Cystinosis can be diagnosed clinically, particularly with the finding of corneal crystals on slit-lamp examination. The diagnosis may be confirmed on laboratory testing with an elevated serum leukocyte cystine measurement.
| 3.0 – 23.0 nmol half-cystine/mg protein | Nephropathic cystinosis |
1.0 – 3.0 nmol half-cystine/mg protein |
Non-nephropathic cystinosis |
≤ 1.0 nmol half-cystine/mg protein |
Heterozygotes for CTNS mutation |
≤ 0.2 nmol half-cystine/mg protein |
Normal value |
Cystinosis is treated with daily cysteamine, which reacts with cystine inside the lysosome to convert it to both cysteine and a cysteine-cysteamine disulfide resembling lysine. These molecules can then be transported out of the lysosome (1,12) Oral cysteamine does improve symptoms and delay organ damage, but end-stage renal disease often still results. Oral cysteamine has also been shown to prevent retinopathy; therefore, it is recommended that all patients begin oral cysteamine therapy at the time of diagnosis (11). Systemic cysteamine has no effect on corneal crystal deposition, but cysteamine eye drops are very effective at dissolving corneal and conjunctival crystals and relieving the associated symptoms when used frequently (every 1-2 hours while awake) (1,13). Genetic testing and prenatal counseling are available.
Epidemiology
|
Signs
|
Symptoms
|
Treatment
|
>Myers HI, Haugsdal JM, Wagoner MD. Cystinosis: A 12-year-old boy with light sensitivity. March 4, 2015; Available from: https://eyerounds.org/cases/209-Cystinosis.htm

Ophthalmic Atlas Images by EyeRounds.org, The University of Iowa are licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License.

Address
University of IowaLegal
Related Links
