
Current Complaint: Blurry vision and tearing
HPI: An 18-year-old male presented for evaluation of suspected aniridia with corneal pannus involving both eyes. His vision had been blurry for years and seemed worse in the left eye. In addition, he had significant watering of the left eye, which he attributed to a scratch to his eye while playing basketball 4 months prior.
Past Ocular History: Refractive error
Past Medical and Surgical History: Non-contributory
Medications: None
Family History: Mother, maternal grandmother, maternal uncle, and possible maternal aunts with aniridia.
Social History: Patient is a former smoker
 |
 |
 |
 |
 |
 |
 |
 |
 |
 |
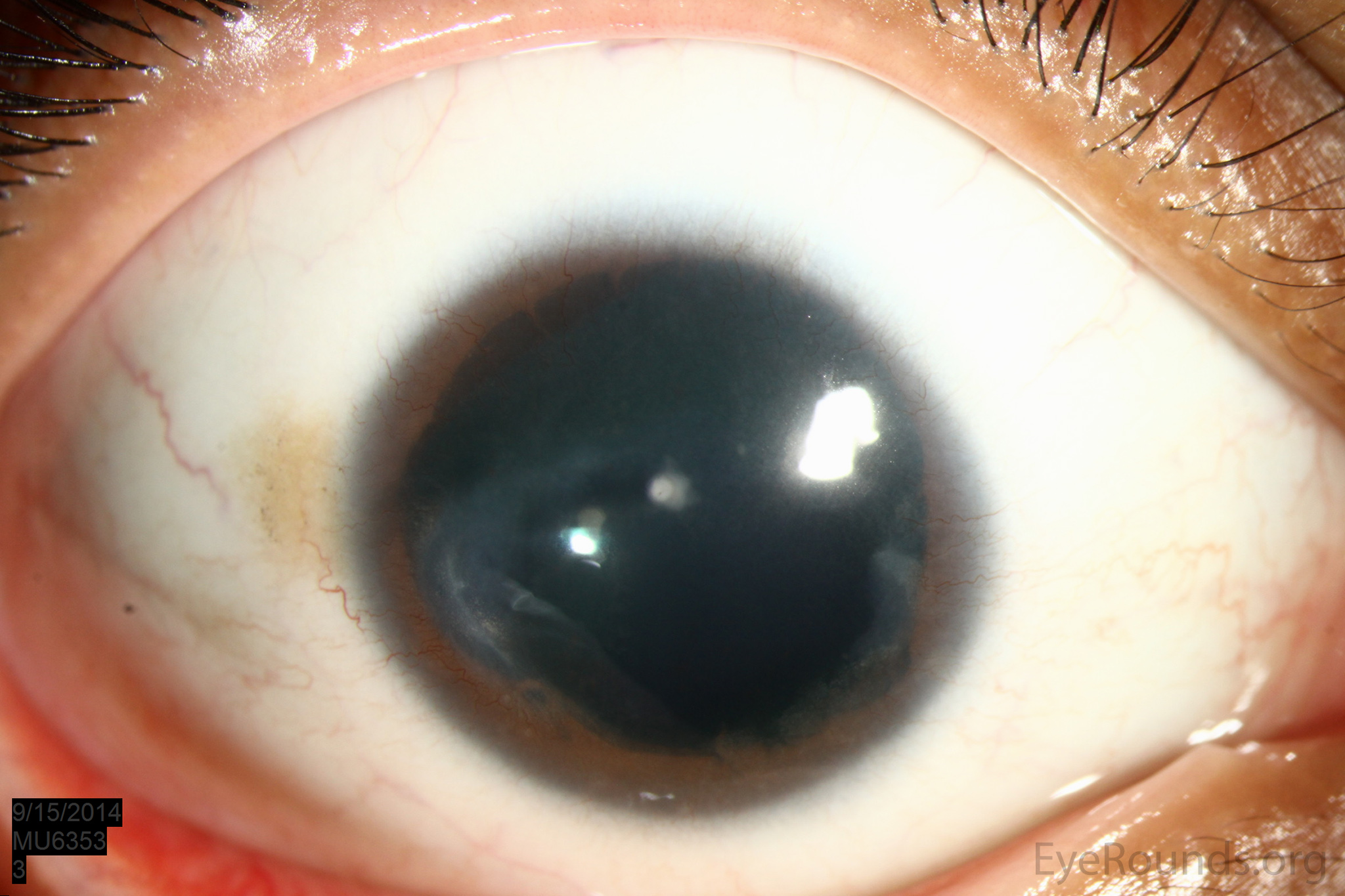
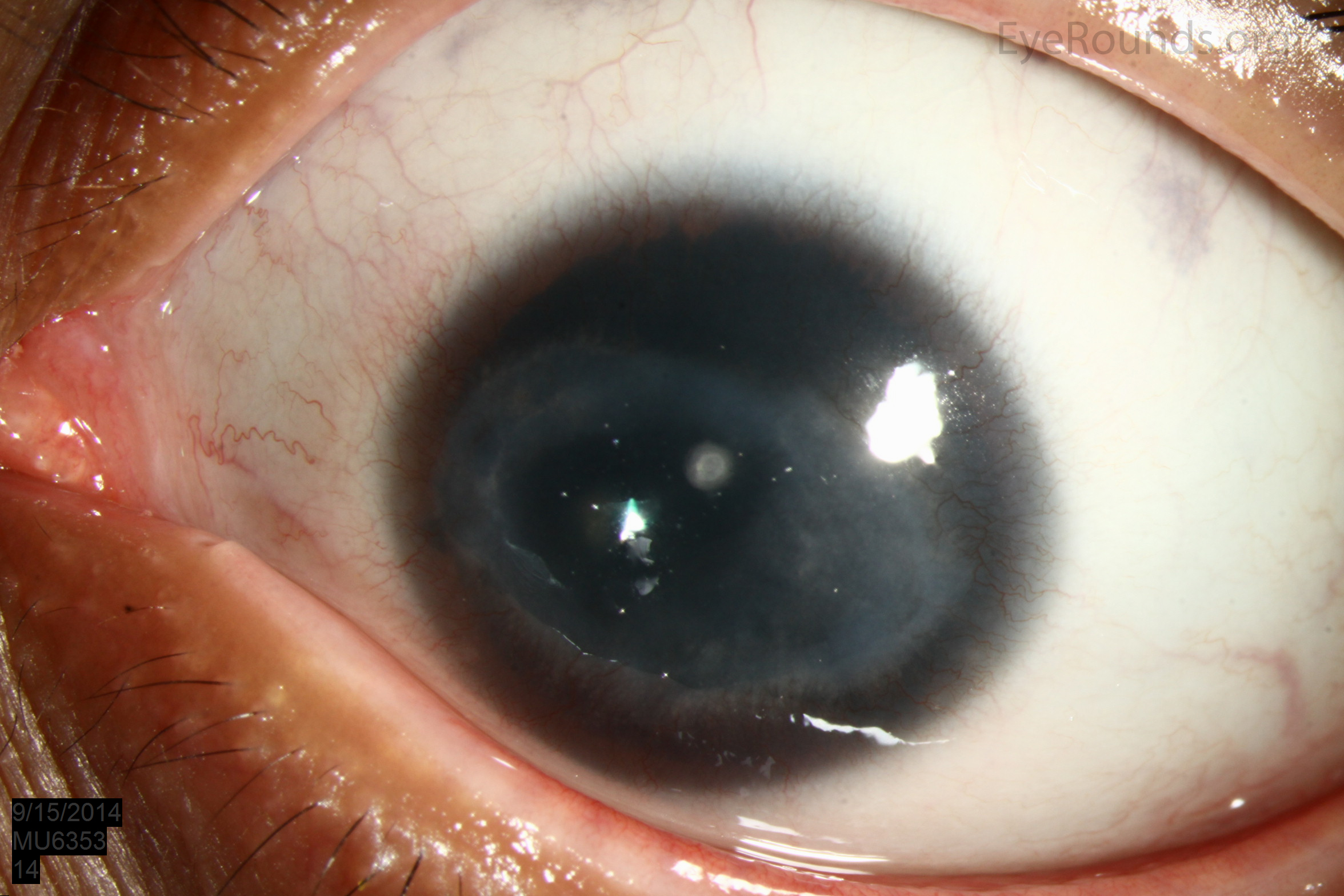
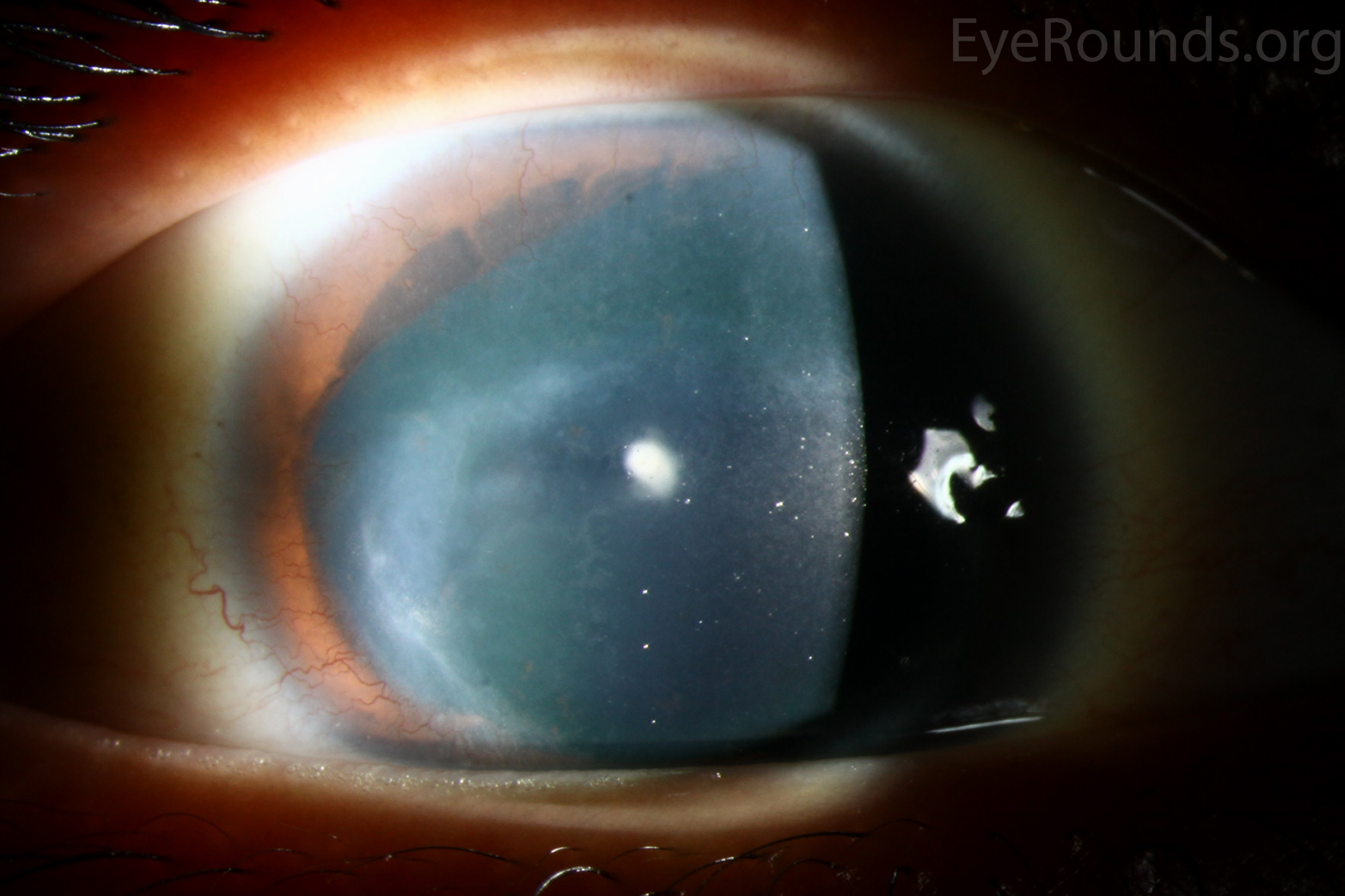
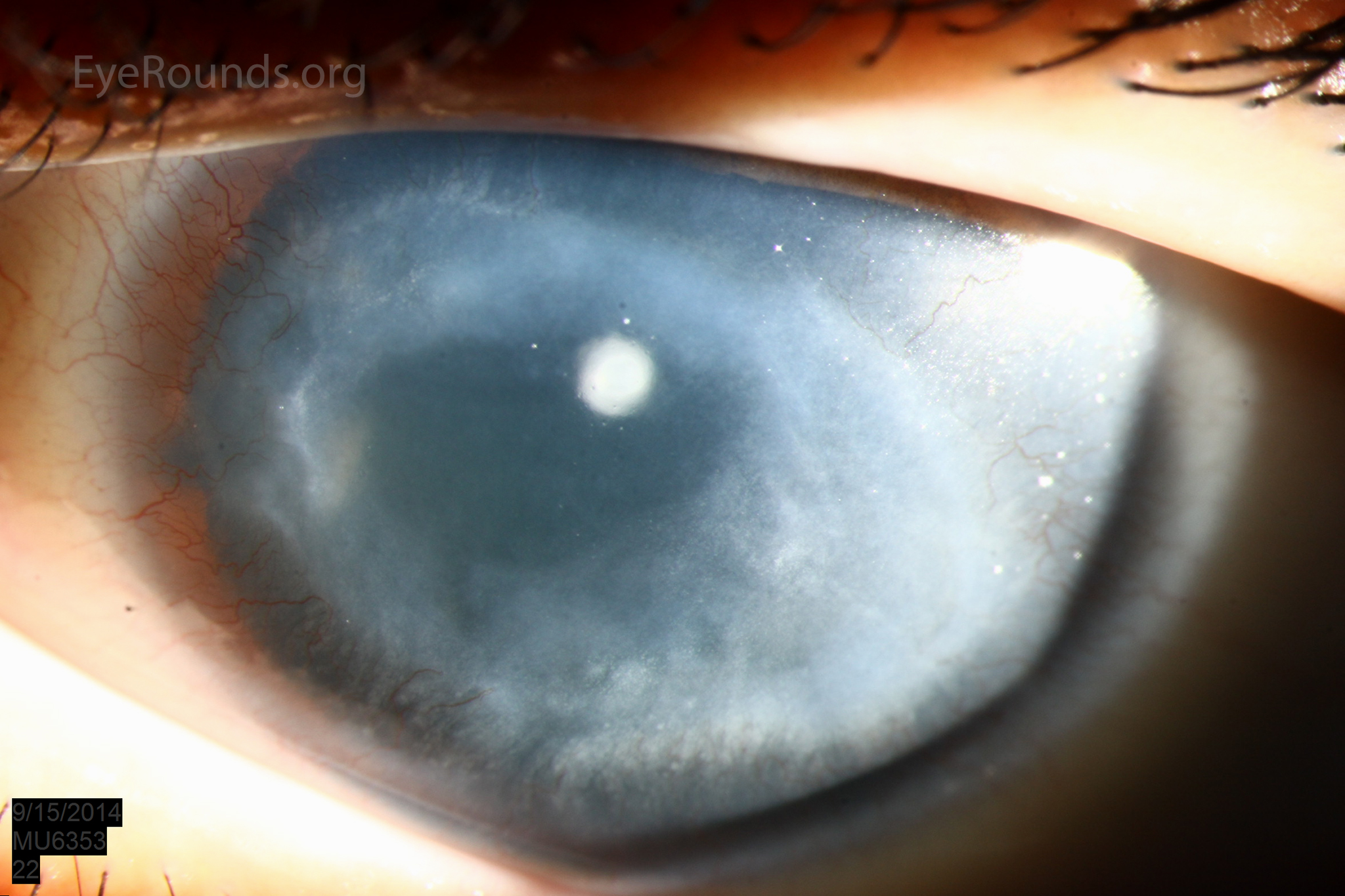
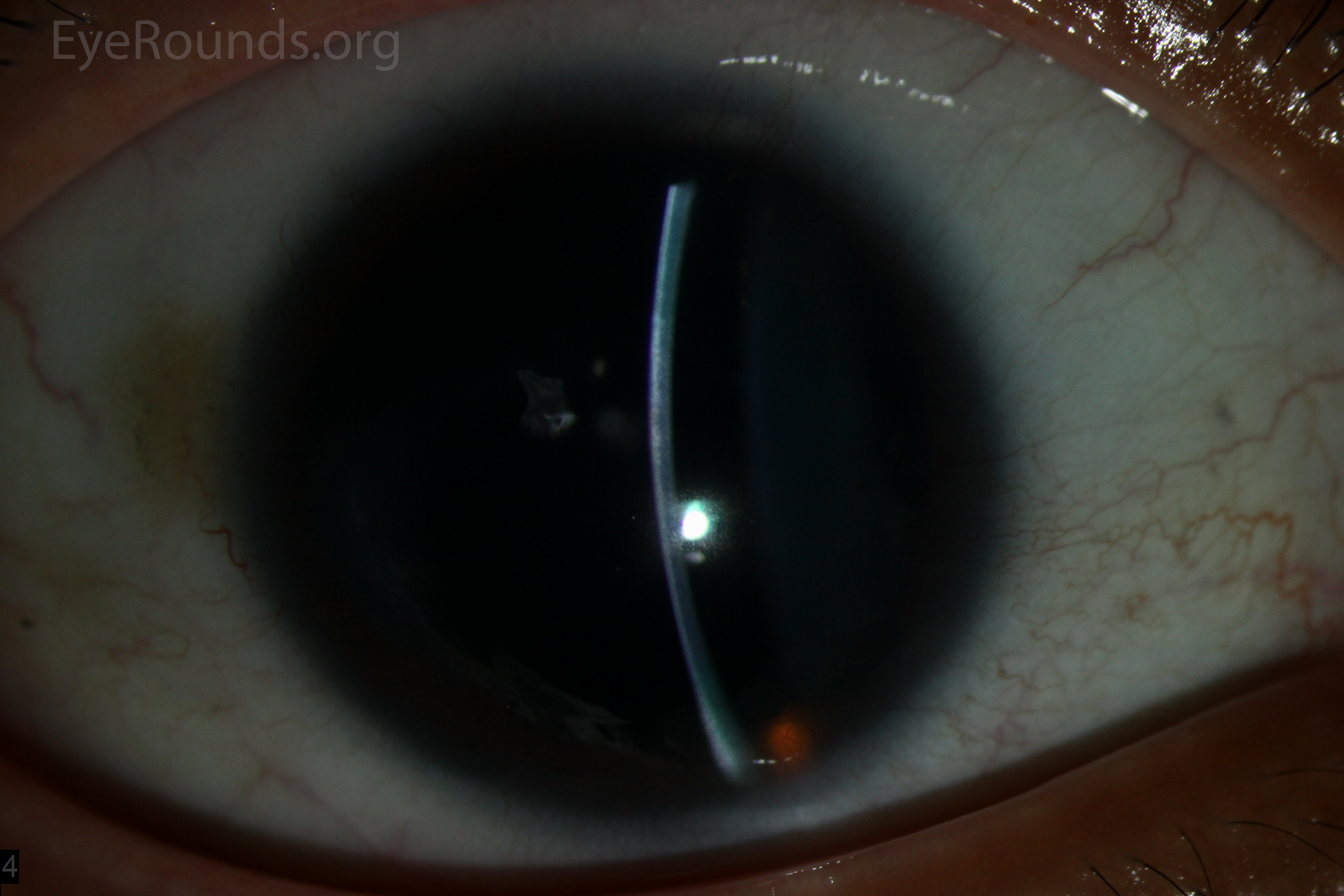
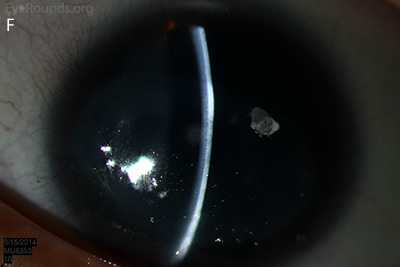
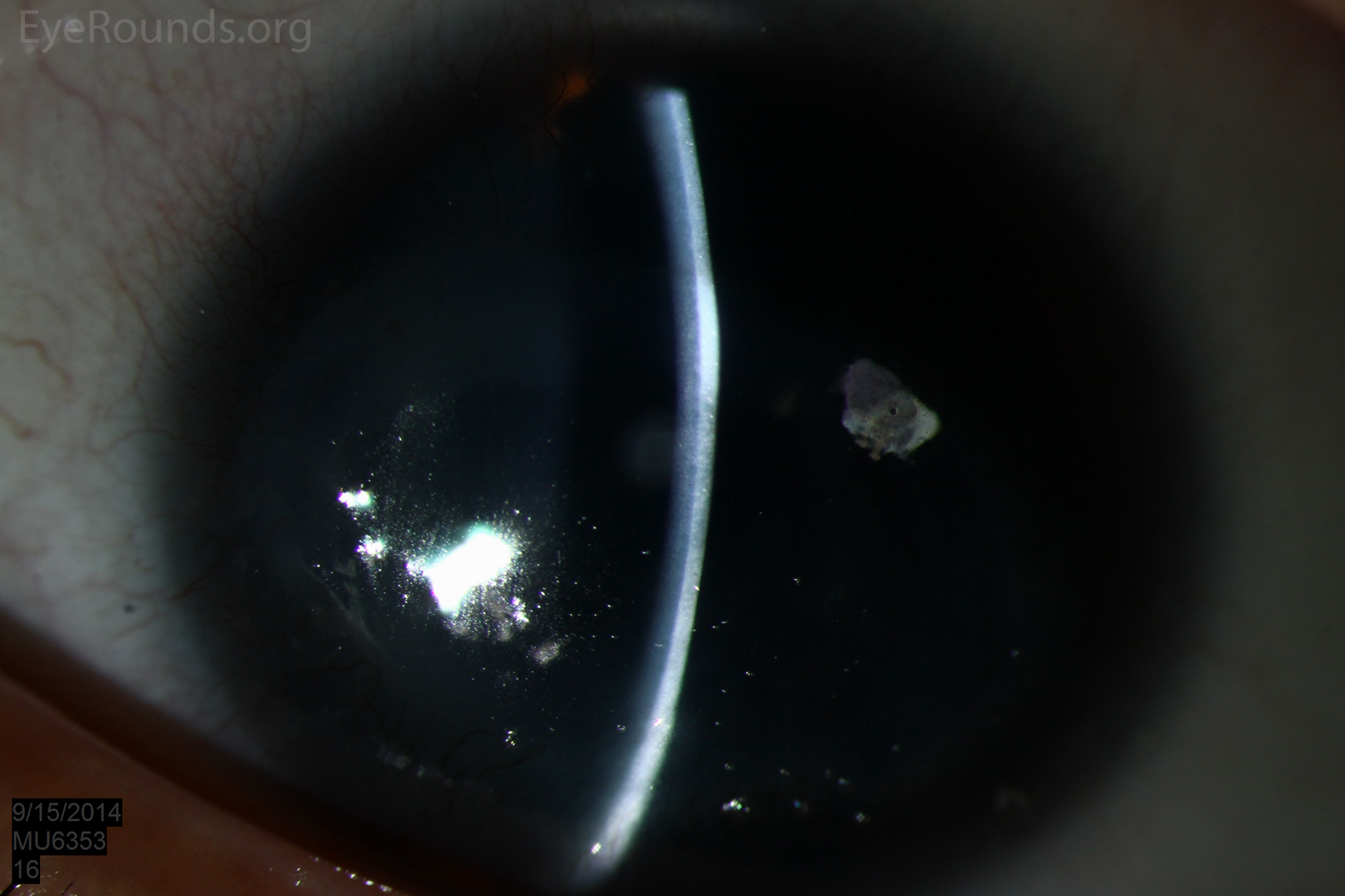
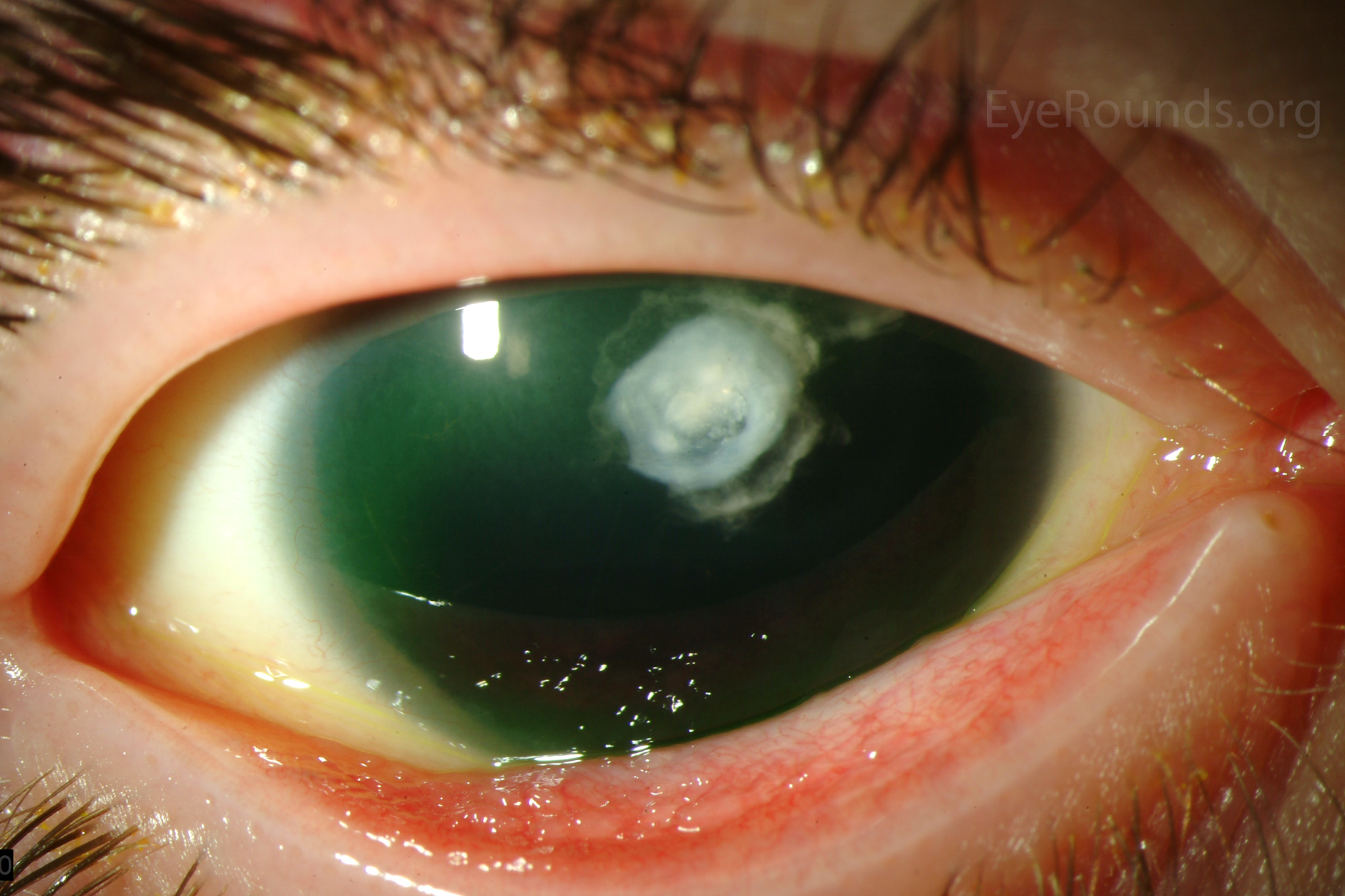
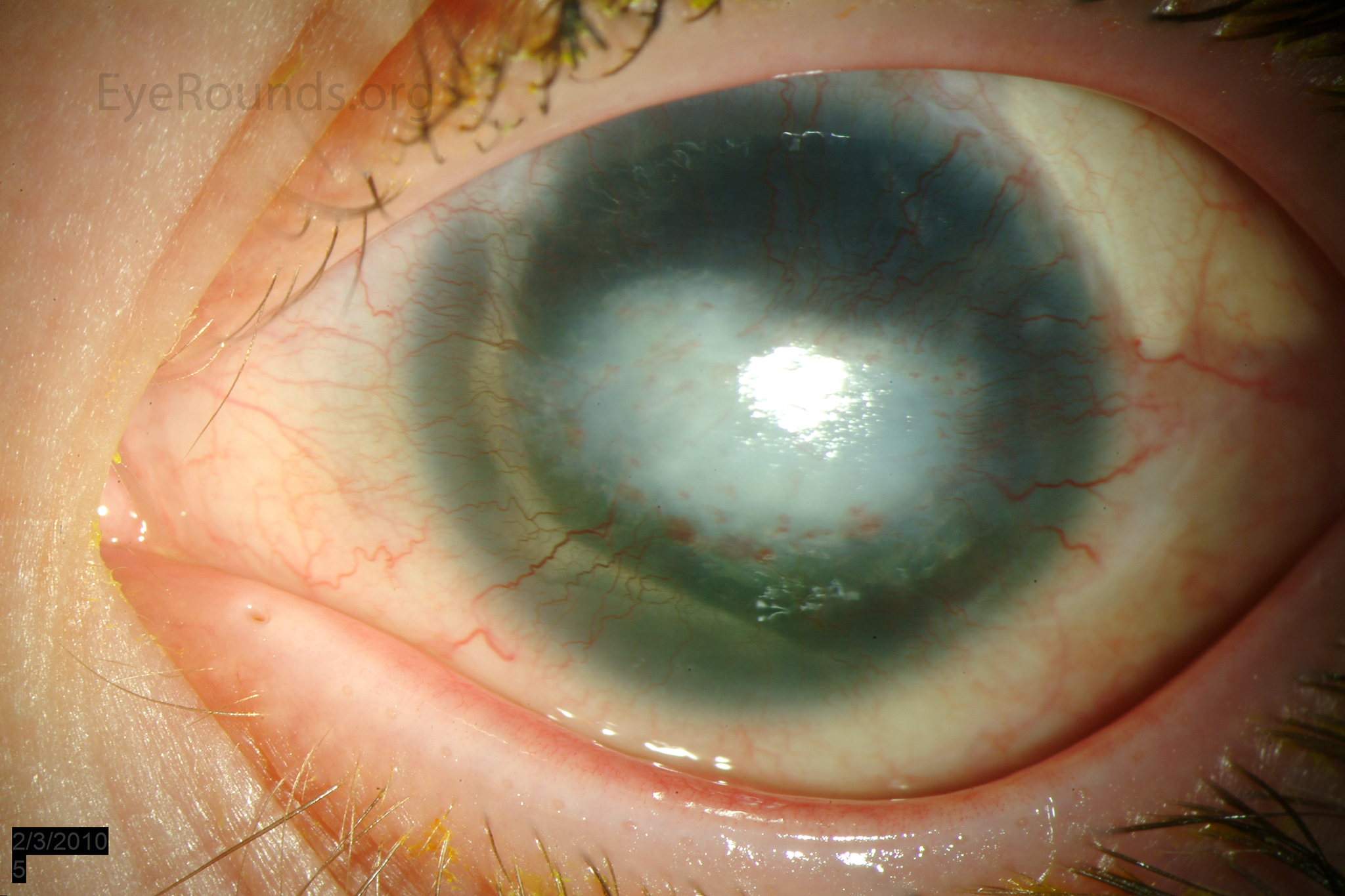
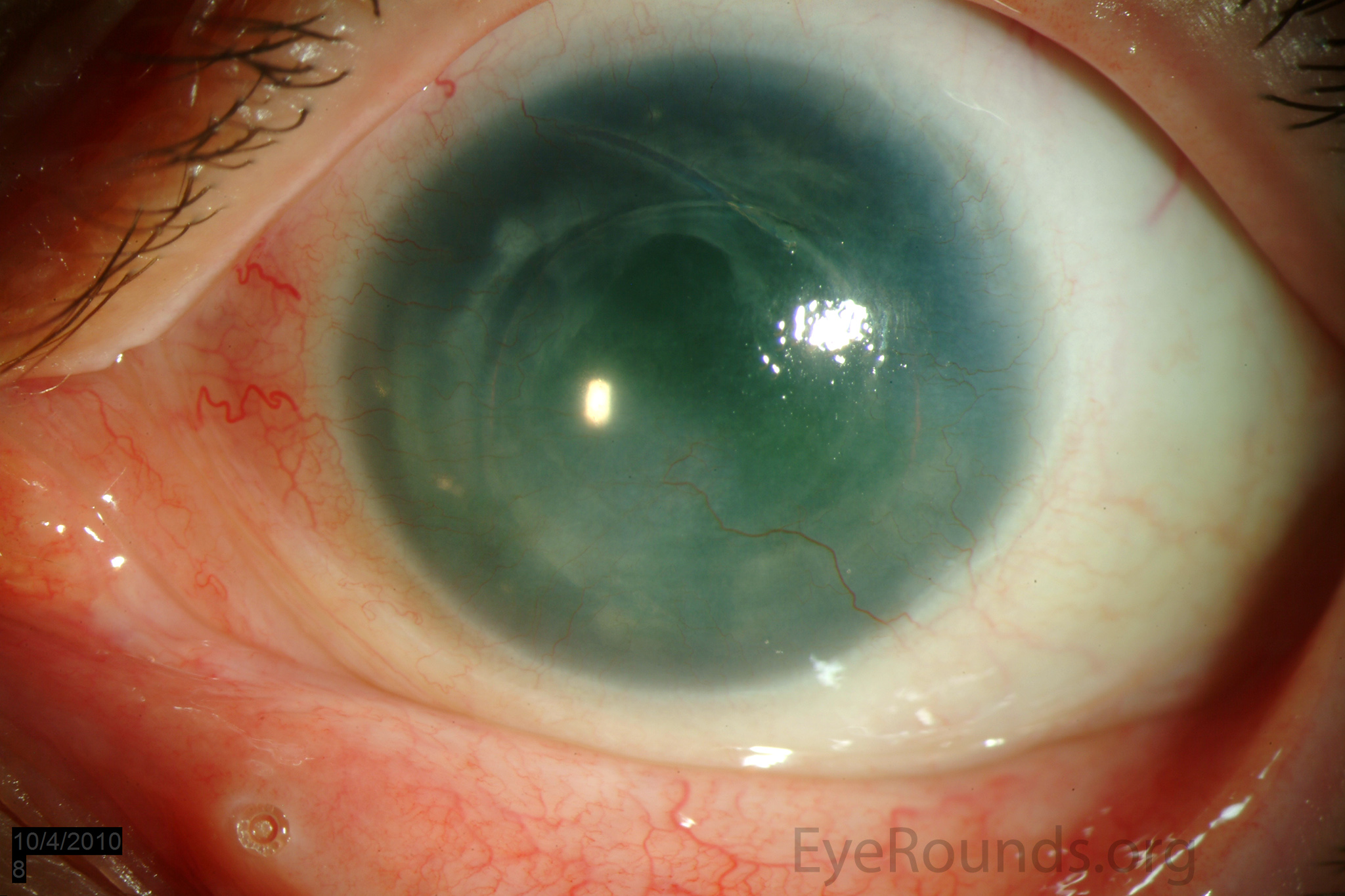
Figure 1: Slit lamp photographs (A, B, C, D, E, F) show corneal pannus, subepithelial haze, and iris hypoplasia. There is fluorescein uptake of the pannus (G, H) due to leaky intracellular junctions. The patient had small anterior polar cataracts (I, J).
The patient's exam was consistent with the diagnosis of aniridia with limbal stem cell deficiency affecting the left eye more than the right and anterior polar cataracts in both eyes. Optical coherence tomography (OCT) of the macula showed poorly defined foveal depression in both eyes consistent with foveal hypoplasia (Figure 2). The cataracts were not felt to be visually significant and were observed. The patient was started on preservative-free artificial tears 6 times daily and lubricating ointment at bedtime in both eyes. The patient was advised to return for follow up in six months. If medical therapy fails, future options include scleral contact lenses, keratolimbal allograft, or keratoprosthesis.
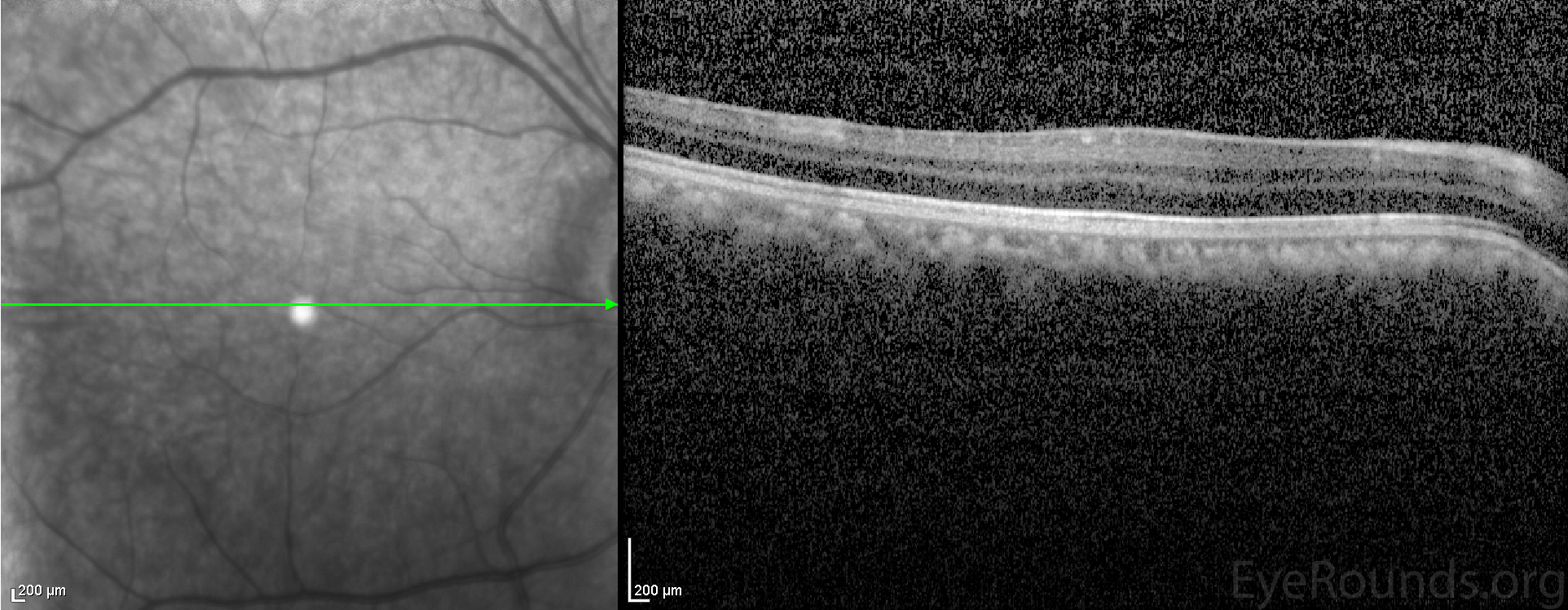
Figure 2: Macular OCT revealed an absence of the normal foveal depression, consistent with foveal hypoplasia.
Congenital aniridia
Aniridia is described as complete or partial absence of the iris. The incidence of the disease is estimated to be between 1: 64,000 and 1:100,000. Isolated aniridia is most often inherited in an autosomal dominant fashion with variable expressivity, however as many as 1/3 of patients inherit the disease sporadically [1]. Aniridia is caused by mutations in the Paired Box gene 6 (PAX6), which is located on chromosome 11p13 and is necessary for the production of protein involved in the early development of the eyes, central nervous system, and pancreas. Over 280 different mutations to the PAX6 gene have been found to cause aniridia, most of which create a premature stop codon leading to the production of nonfunctional protein [2]. When it occurs sporadically, aniridia may also occur as a part of the WAGR syndrome (Wilms tumor, aniridia, genitourinary anomalies, and mental retardation). This syndrome is caused by a deletion of the short arm of chromosome 11, with the loss of both the PAX6 and WT1 genes. WT1 is necessary for the production of a protein involved in the development of the kidneys and gonads [3].
Aniridia is a bilateral disease associated with multiple ocular abnormalities (Figure 3). Characteristics of the disease include varying degrees of iris hypoplasia, limbal stem cell deficiency and corneal opacification, cataracts, foveal hypoplasia, optic nerve hypoplasia, glaucoma, nystagmus, and decreased visual acuity [4]. The term aniridia is a misnomer, as all patients have at least a rudimentary iris stump, though it may only be visible on gonioscopy. The cornea can be affected by aniridia-associated keratopathy secondary to limbal stem-cell deficiency. Signs of keratopathy include thickening and vascularization of the peripheral cornea (pannus), which can advance to involve the entire cornea. This leads to corneal erosion, ulceration, and fibrosis with opacification. Symptoms of aniridia-associated keratopathy include dryness, erythema, photophobia, and epiphora. Meibomian gland dysfunction and an abnormal tear film may also be present [1]. Congenital cataracts may be mild at birth, but can advance through childhood and early adulthood to become more visually symptomatic. Foveal hypoplasia is a potentially devastating sequela of aniridia resulting in decreased vision, often with associated nystagmus, from birth. Optic nerve hypoplasia occurs in about 10% of individuals and can also affect vision. Glaucoma develops in 1/2 to 2/3 of patients with aniridia. Patients may have glaucoma at a young age secondary to abnormal iridocorneal angle development, but most develop glaucoma later in childhood or early adulthood. The mechanism of glaucoma is believed to be secondary to the rudimentary iris stump folding anteriorly over the trabecular meshwork [5]. Symptoms vary between affected individuals, but rarely is there variation between an individual's two eyes [4]. Slit lamp examination, fundoscopy, optical coherence tomography, high-frequency ultrasound biomicroscopy and iris fluorescein angiography may all be used to detect the clinical features of aniridia [6].
 |
 |
 |
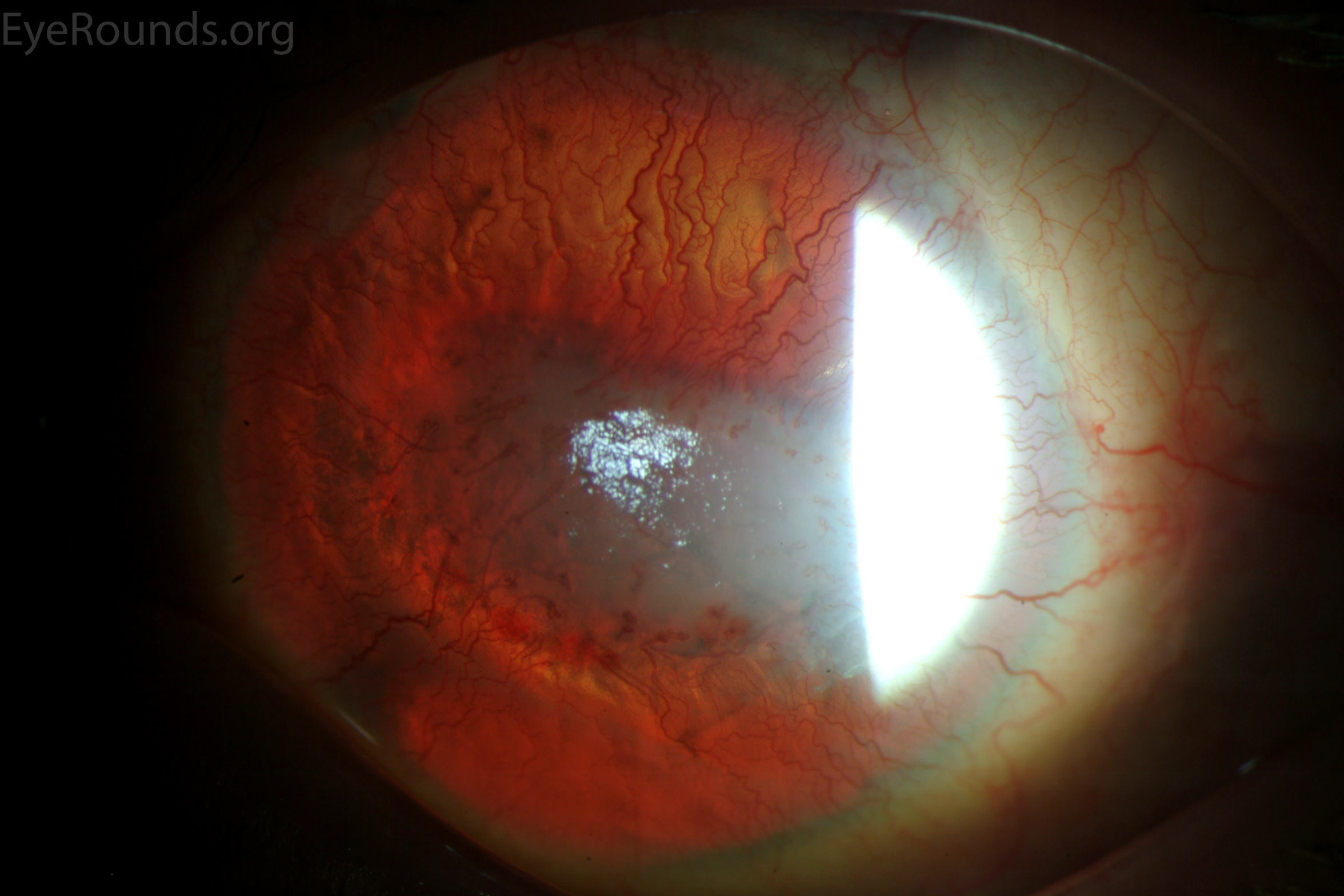 |
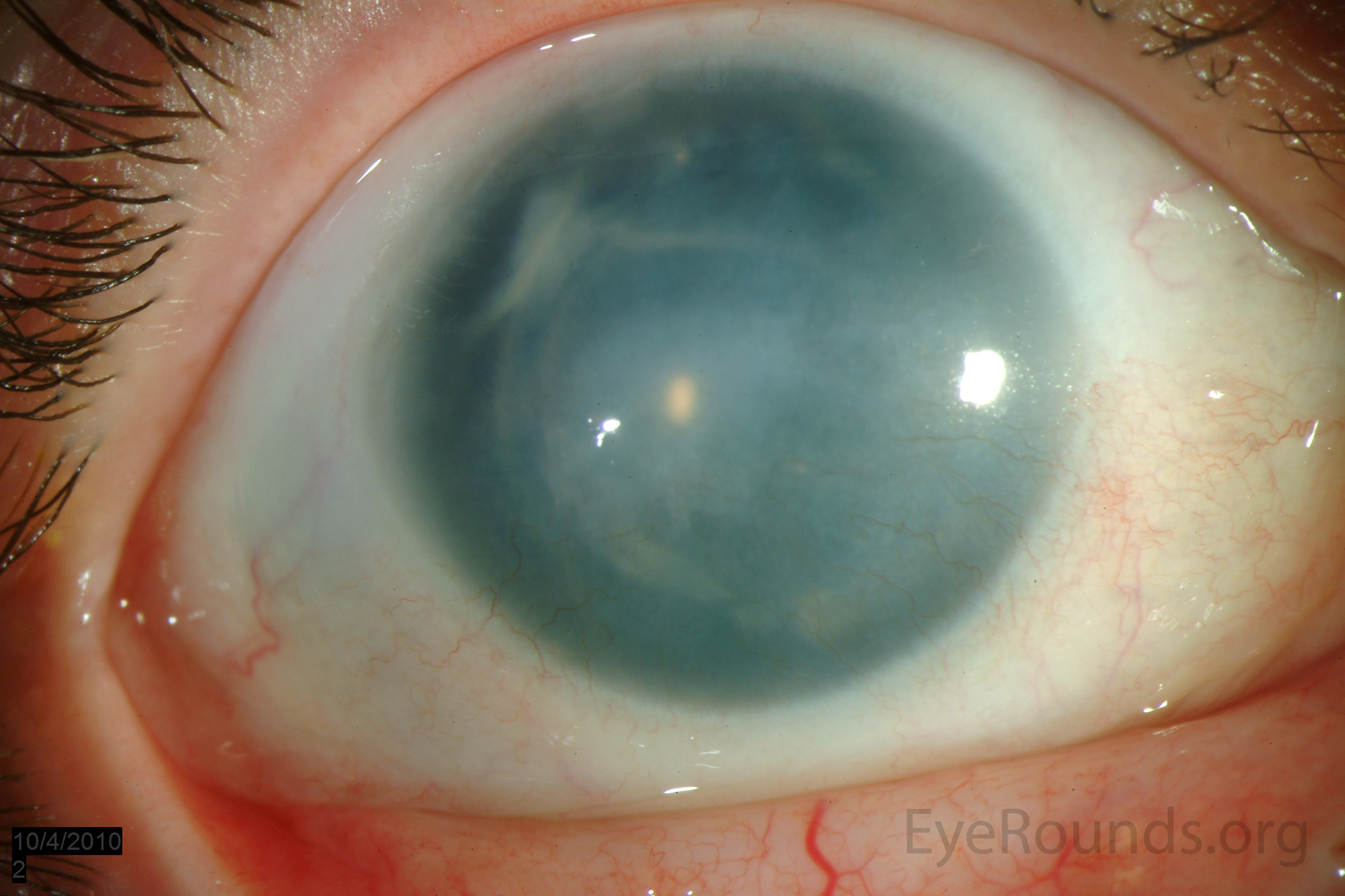
Figure 3: Slit lamp photographs (A, B) of another patient with congenital aniridia. There is conjunctivalization of the left cornea due to stem cell deficiency and a central corneal scar. There is iris hypoplasia in both eyes. The patient has a congenital cataract in the right eye and is aphakic in the left eye after having undergone cataract surgery. The cataract and corneal scars are demonstrated on retroillumination (C, D). Also note the visible inferior border of the crystalline lens and zonules in the right eye, a feature not visible in individuals with normal irides.
 |
 |
 |
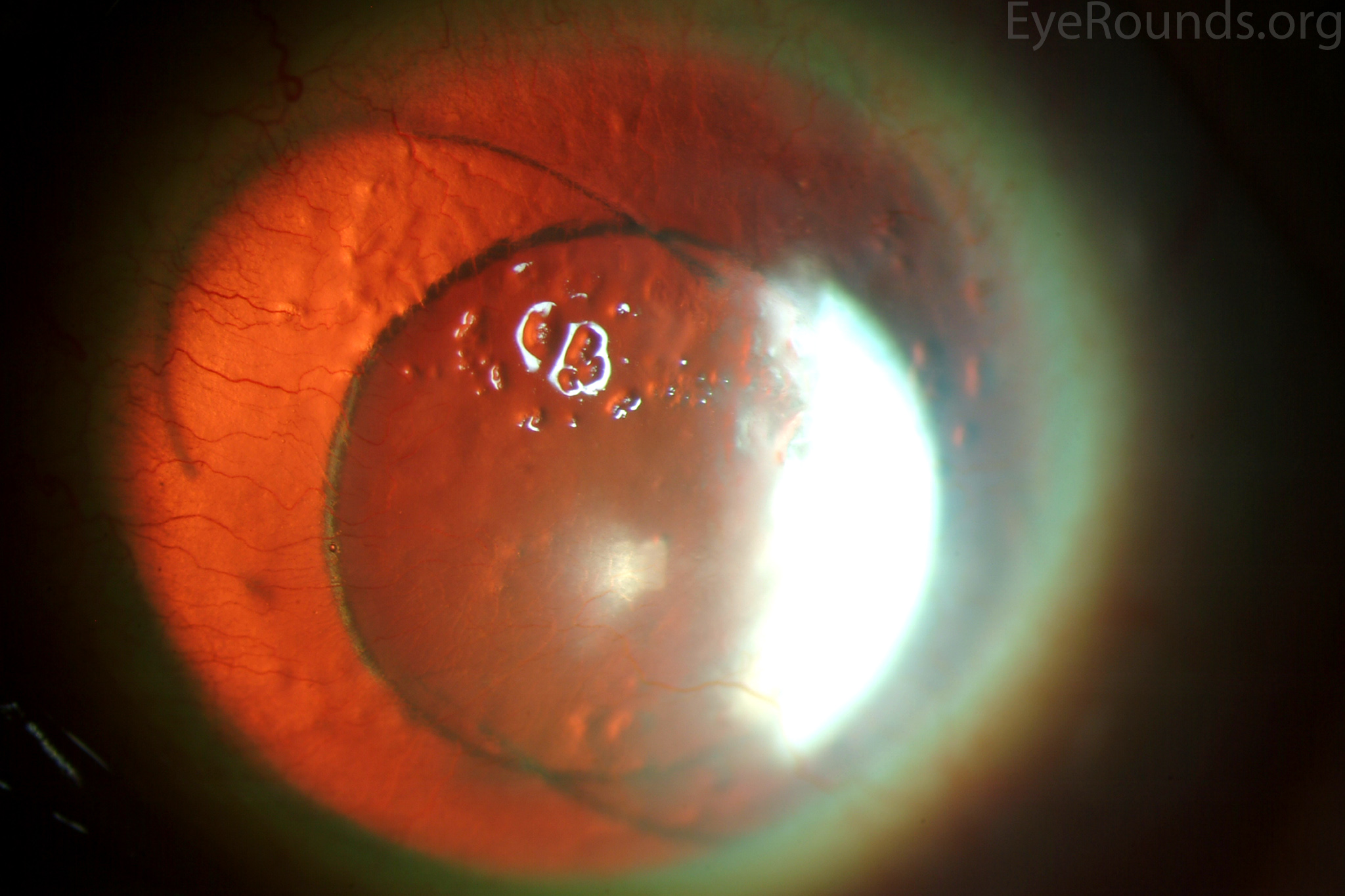 |
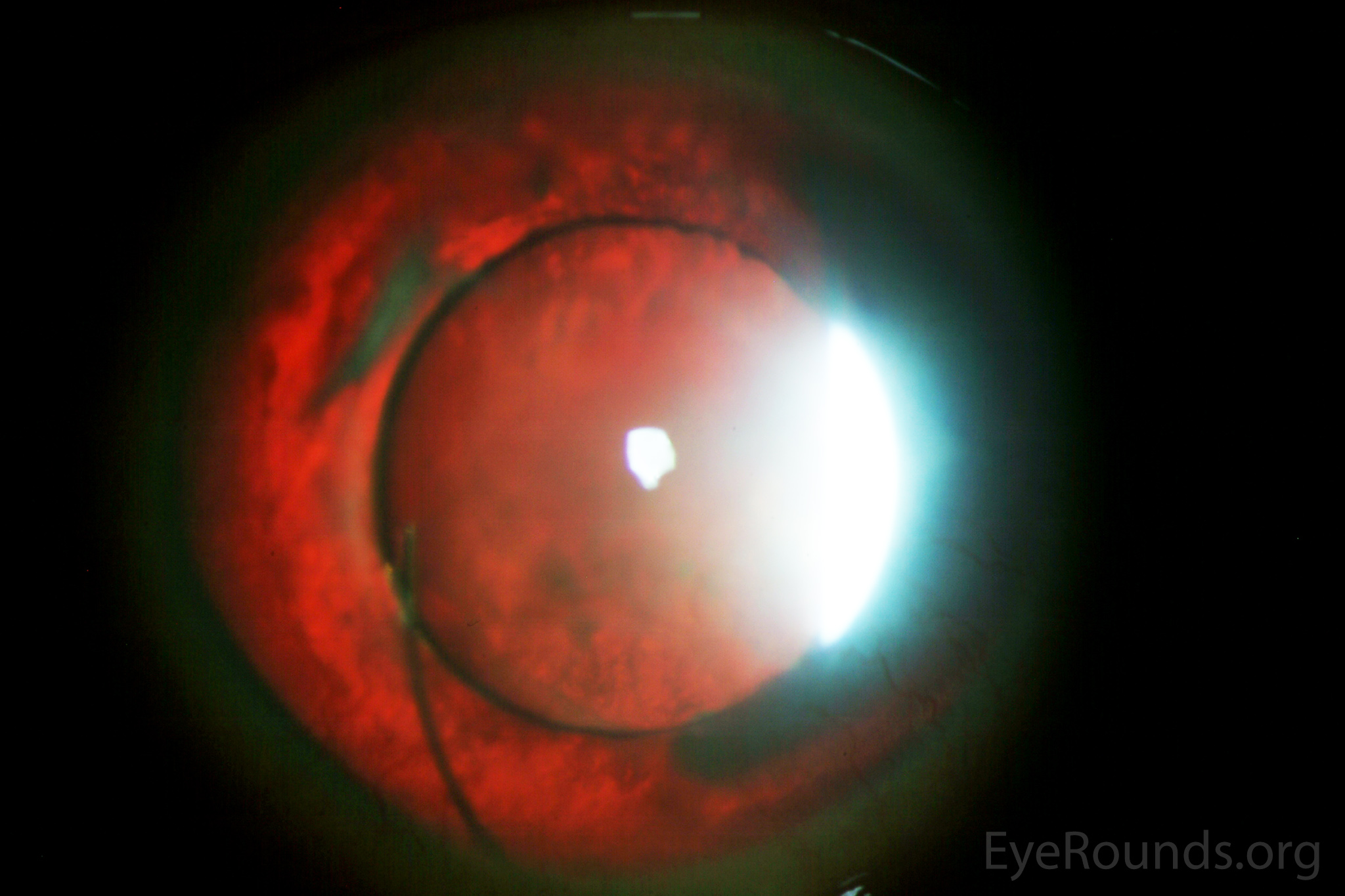
Figure 4: This patient with congenital aniridia has 3-piece posterior chamber intraocular lenses in both eyes (A, B). They are seen on retroillumination (C, D).
Aniridia is a clinical diagnosis, but confirmational genetic testing is available. Once a clinical diagnosis has been made, it is important to evaluate for a family history of the disease. If there is a positive family history, especially in a parent of the affected patient, it is unlikely that the patient will have a deletion of WT1 causing WAGR syndrome. Sporadic aniridia must be evaluated for the deletion of WT1, since there is a high risk of pediatric nephroblastoma (Wilms tumor). The first step in genetic testing is to evaluate for the presence of a genomic deletion with high resolution CGH array. If only a PAX6 deletion is present, the diagnosis of isolated aniridia is confirmed. If both PAX6 and WT1 deletions are detected, WT1 deletion should be confirmed through further testing, such as FISH. WT1 deletion carriers require frequent renal ultrasound monitoring for early detection of Wilms tumor. If no deletion is detected through CGH, sequence analysis may be done to evaluate for intragenic mutations within PAX6. If the test is still negative, the patient may have a mutation in aniridia-associated genes, such as PITX2, PITX3, and FOXC1 (transcription factors involved in the development of the eye) [4].
Treatment of aniridia often involves management of multiple pathologic processes. Initial care is often conservative and patients should have regular eye examinations including screening for glaucoma and optic nerve changes. Refractive error can be corrected with glasses with tinted lenses to help reduce light sensitivity [4].
Ocular surface dysfunction may initially be treated with preservative-free artificial tears, lubricating ointment and punctal occlusion [6]. With severe corneal disease, surgical management, such as keratolimbal allograft (KLAL), may be necessary (Video 1). Penetrating keratoplasty has a poor success rate due to limbal stem cell deficiency, which leads to corneal neovascularization and graft failure [1]. Holland et al. showed that keratolimbal allograft is effective in treating aniridic keratopathy, especially in those receiving systemic immunosuppression. The Boston keratoprosthesis (KPro) has been increasingly used, especially in patients with previous graft failure (Figure 5) and has been shown to be effective in significantly improving visual acuity [8].
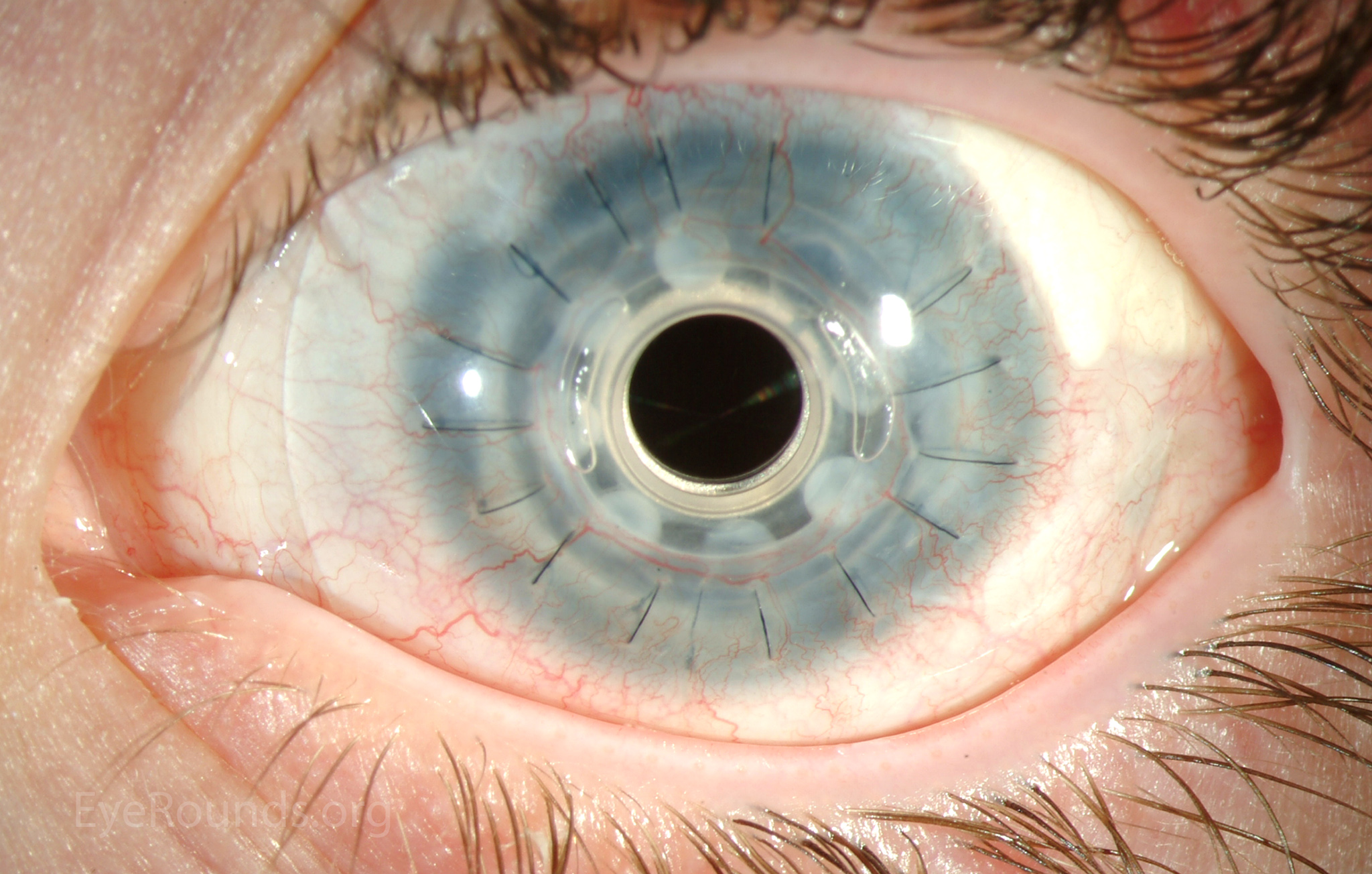
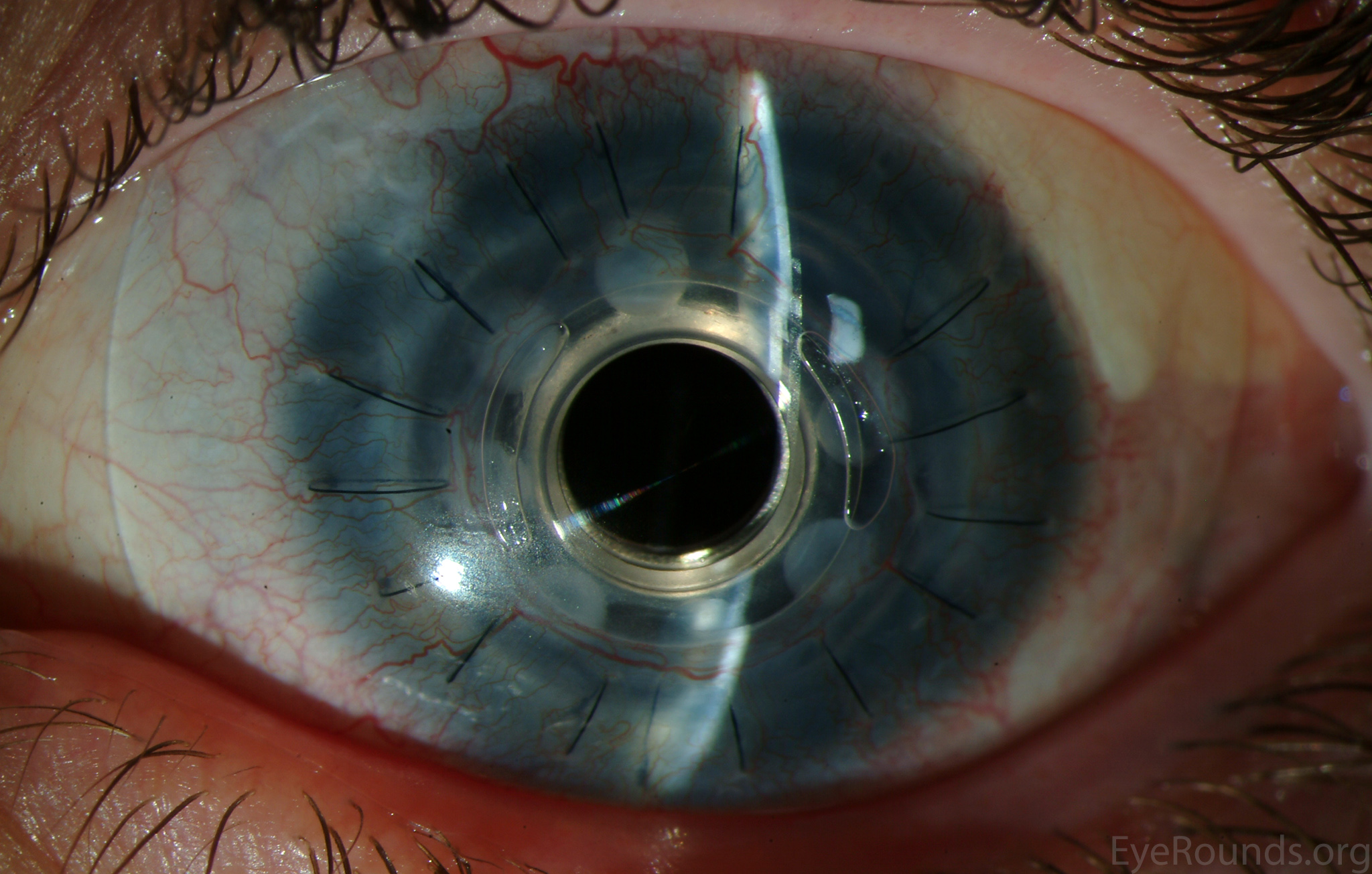
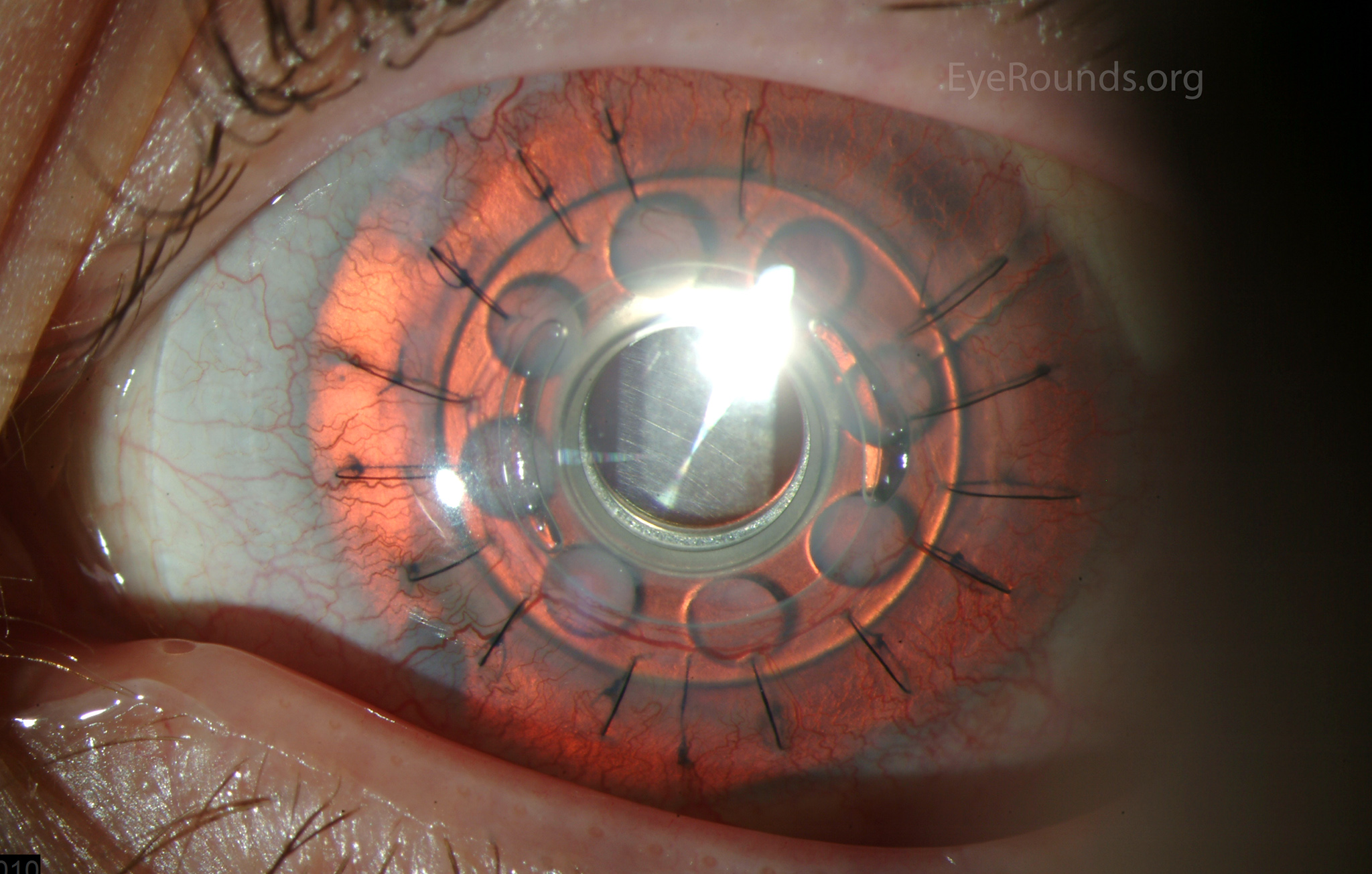
Figure 5: Postoperative appearance of the patient in Figure 3 after placement of a Boston keratoprosthesis (KPro).
Video: Keratolimbal allograft surgery
Several intraocular implants are available to simulate the appearance of an iris, though none are currently FDA approved in the United States. Morcher makes a variety of intraocular implants made of solid black PMMA that can come with or without a clear central refracting lens (Figure 6). They are not as cosmetically attractive as some of the alternatives, but can decrease glare. More recently, Morcher has offered a one-piece PMMA model that comes in 45 different colors. These lenses require a 9-9.5 mm incision for implantation. The Ophtec 311 intraocular lens implant has been used in Europe for several years (Figure 7) and features a colored body and haptics to simulate the appearance of an iris. The lens comes in blue, brown, and green and requires a 9 mm incision for implantation, though the company has an alternate device that is assembled inside the eye to allow the use of a 5 mm incision. Phase I studies in the United States have shown that the Ophtec 311 can improve visual acuity and decrease photophobia and glare [9]. The CustomFlex prosthesis is an artificial iris made of a foldable silicone elastomer (Figure 8). The device is custom-made to match the contralateral eye in patients with unilateral defects or designed from patient selection in the case of bilateral aniridia. The prosthesis is designed to fit in the sulcus or capsular bag and its foldable nature allows it to fit through a 2.75 mm incision, which is an advantage compared to the other iris prostheses. The company recommends that its use be limited to pseudophakic eyes only.
 |
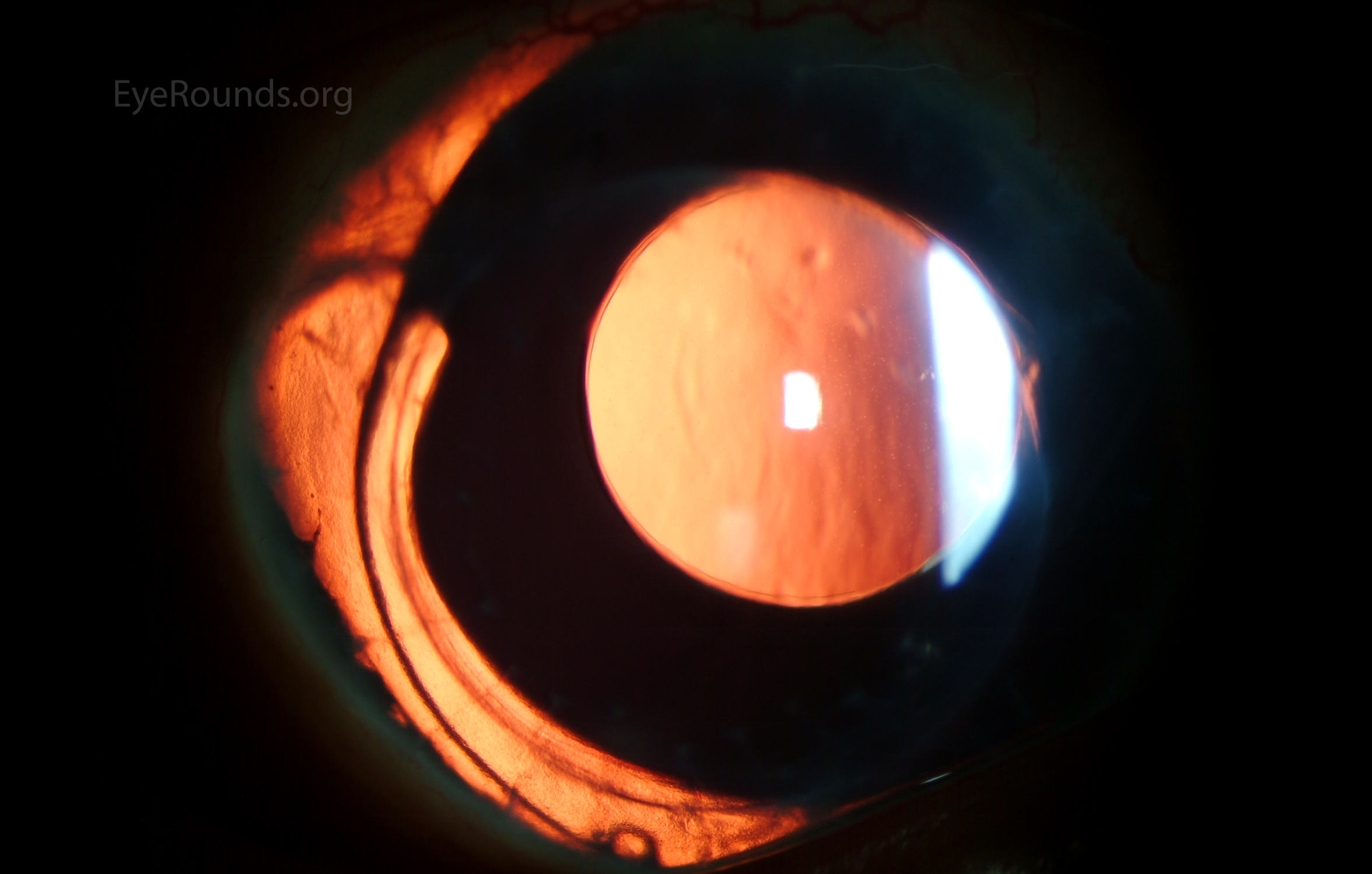 |
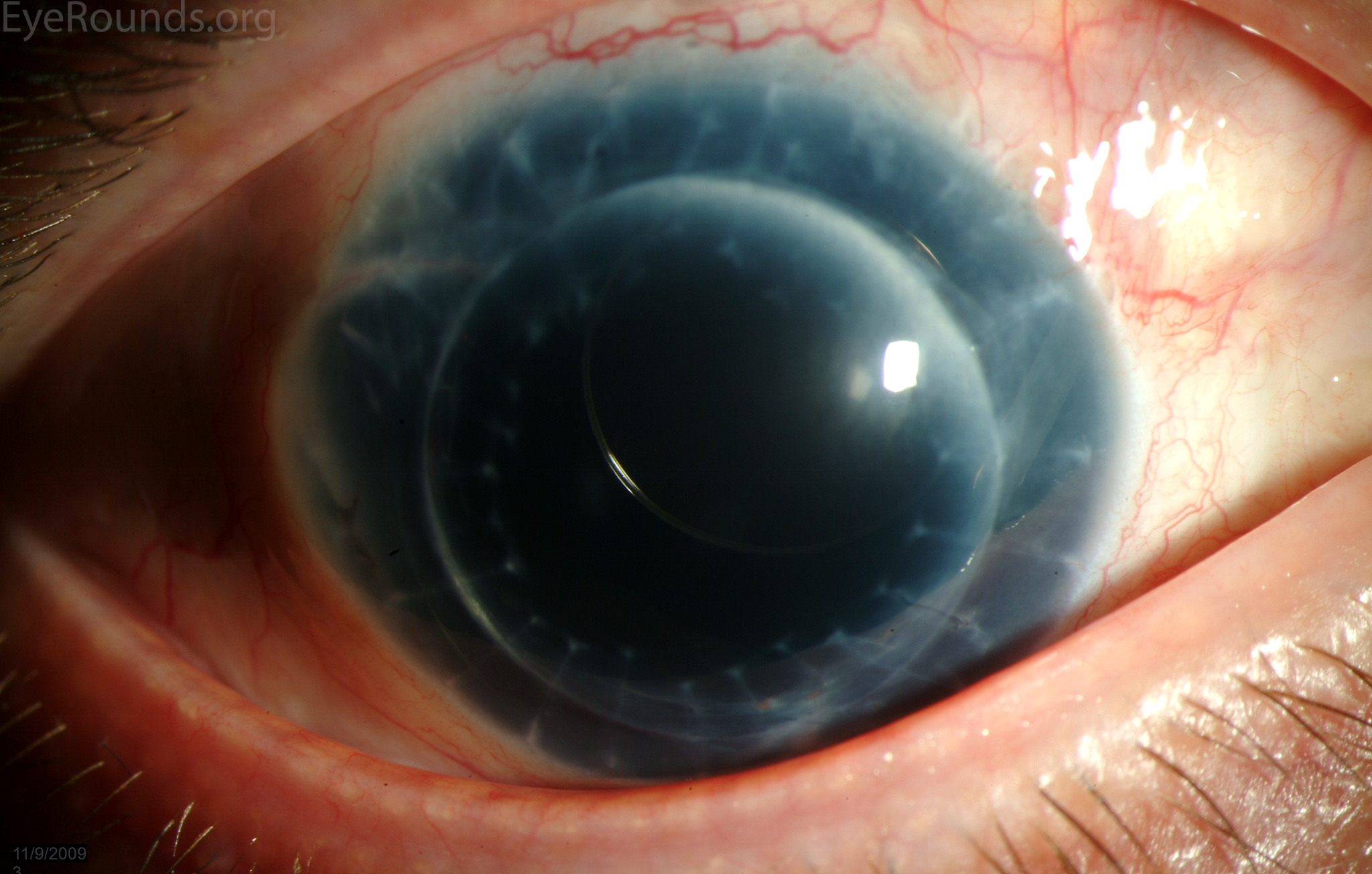
Figure 6: A Morcher intraocular lens in an eye with a full-thickness corneal transplant and traumatic aniridia.
 |
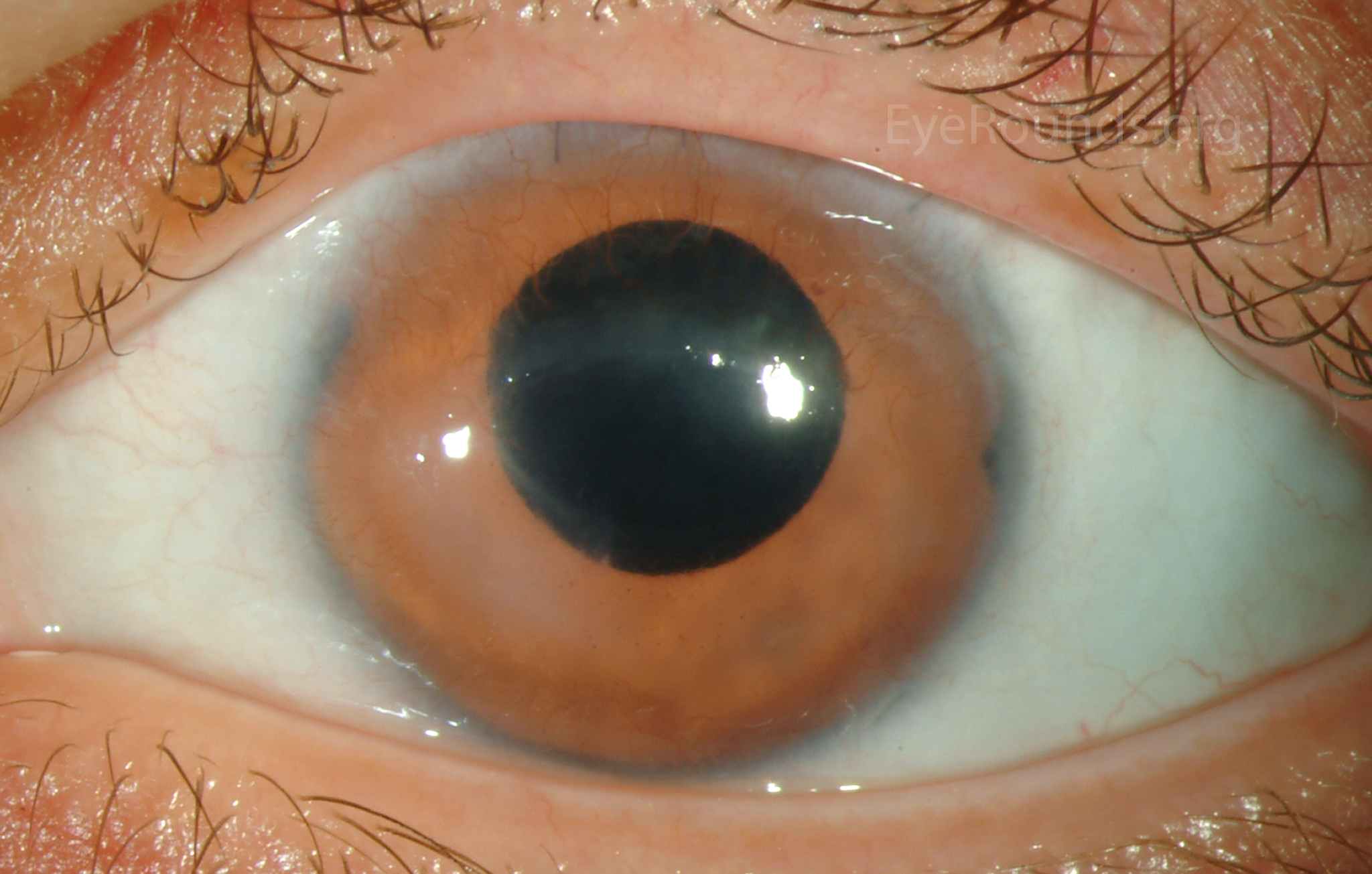 |
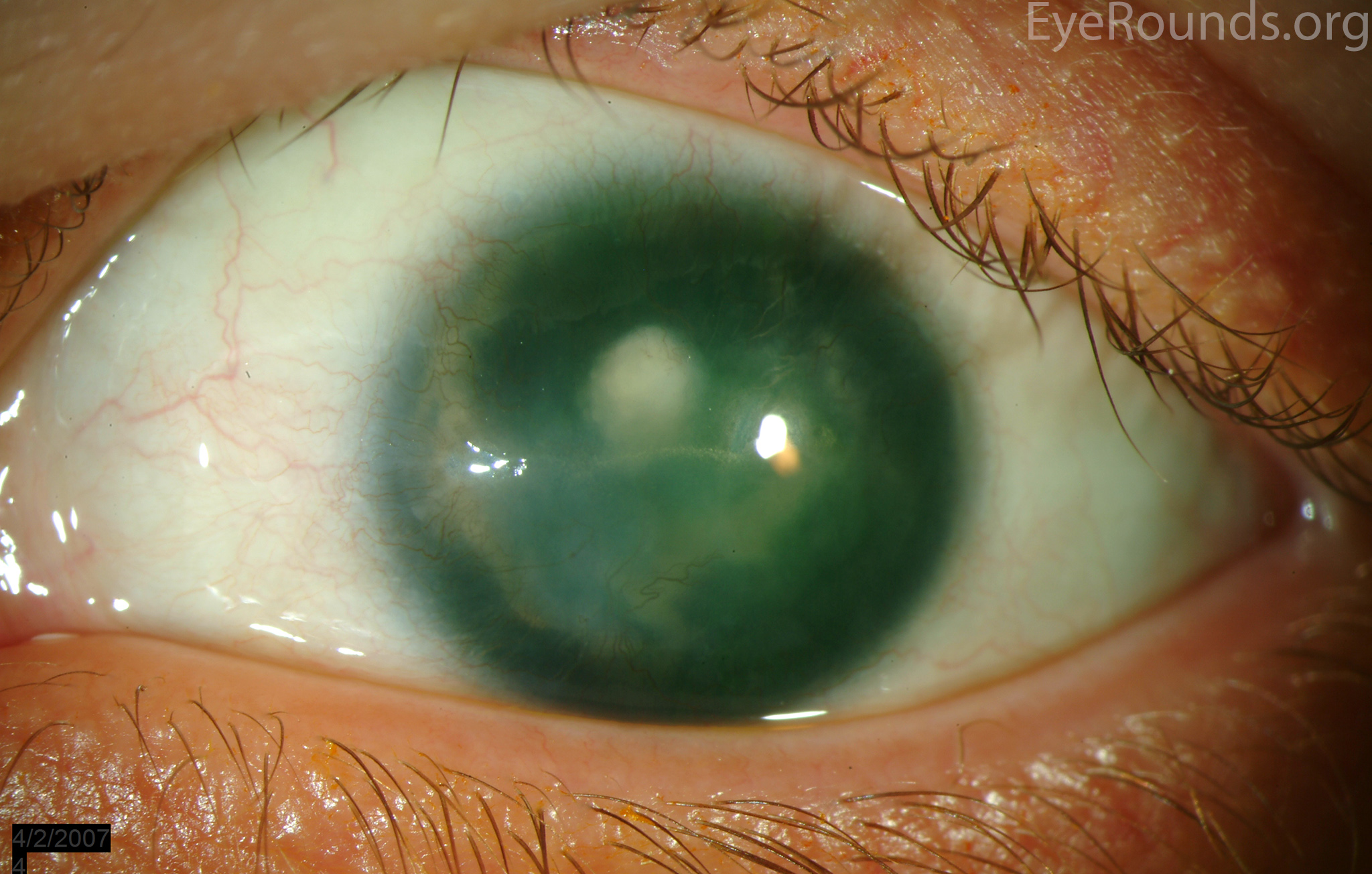
Figure 7: (A) Preoperative appearance of a patient with congenital aniridia and associated cataract. (B) Appearance after cataract extraction and implantation of an Ophtec 311 iris reconstruction intraocular lens with artificial iris.

Figure 8: A CustomFlex iris prosthesis provides an excellent cosmetic result in this patient with history of penetrating keratoplasty for acanthamoeba keratitis and persistent mydriasis.
Glaucoma is often initially treated with topical medications. In very young patients, trabeculotomy or goniotomy can be considered due to abnormal iridocorneal angle development. Ultimately, patients may require further surgical intervention including trabeculectomy, glaucoma drainage device implantation, or cyclodestructive procedures [1].
Cataract formation is common in aniridia. Small cataracts may be observed if they are minimally visually significant. Physicians must be aware that there is increased fragility of the anterior capsule in patients with aniridia to avoid intraoperative complications [1].
Trauma
Post-surgical
Rieger syndrome
Iris coloboma
Gillespie syndrome
Iridocorneal endothelial (ICE) syndrome
Epidemiology
|
Signs
|
Symptoms
|
Treatment
|
Goetz K, Vislisel JM, Raecker ME, Goins KM. Congenital Aniridia. March 10, 2015; Available from: https://eyerounds.org/cases/211-Aniridia.htm

Ophthalmic Atlas Images by EyeRounds.org, The University of Iowa are licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License.

Address
University of IowaLegal
Related Links
