
"My eye is sticking out"
A 43-year-old female was referred to the University of Iowa Oculoplastics Clinic by her comprehensive ophthalmologist for the evaluation of worsening right-sided proptosis. She reported that over the past 20 years her right eye had gradually moved forward. She denied pain, double vision, or tearing. She denied any recent trauma or malignancy but was kicked on the right side of the head by a cow at the age of 10. She reported a history of hypothyroidism. At the age of 36, she had a computed tomography (CT) scan for proptosis by an outside provider that showed an osseous mass in the greater wing of the sphenoid along the superolateral wall of the orbit.
No known history of bone disease or ocular disease
Non-smoker
Negative except for what is detailed in the history of present illness
with correction (Snellen-Linear)
| OD | OS | |
| External | proptotic | normal |
| MRD1 | 6 mm | 5 mm |
| MRD2 | 8 mm | 6 mm |
A CT was performed which showed expansion of the right greater wing of the sphenoid bone consistent with fibrous dysplasia with reduced lateral diameters of the orbit on the right (Figure 1). Because the patient had no visual compromise, her ocular surface was healthy, and she was not interested in surgical intervention, the decision was made to have the patient follow up with her local eye care provider and return as needed.
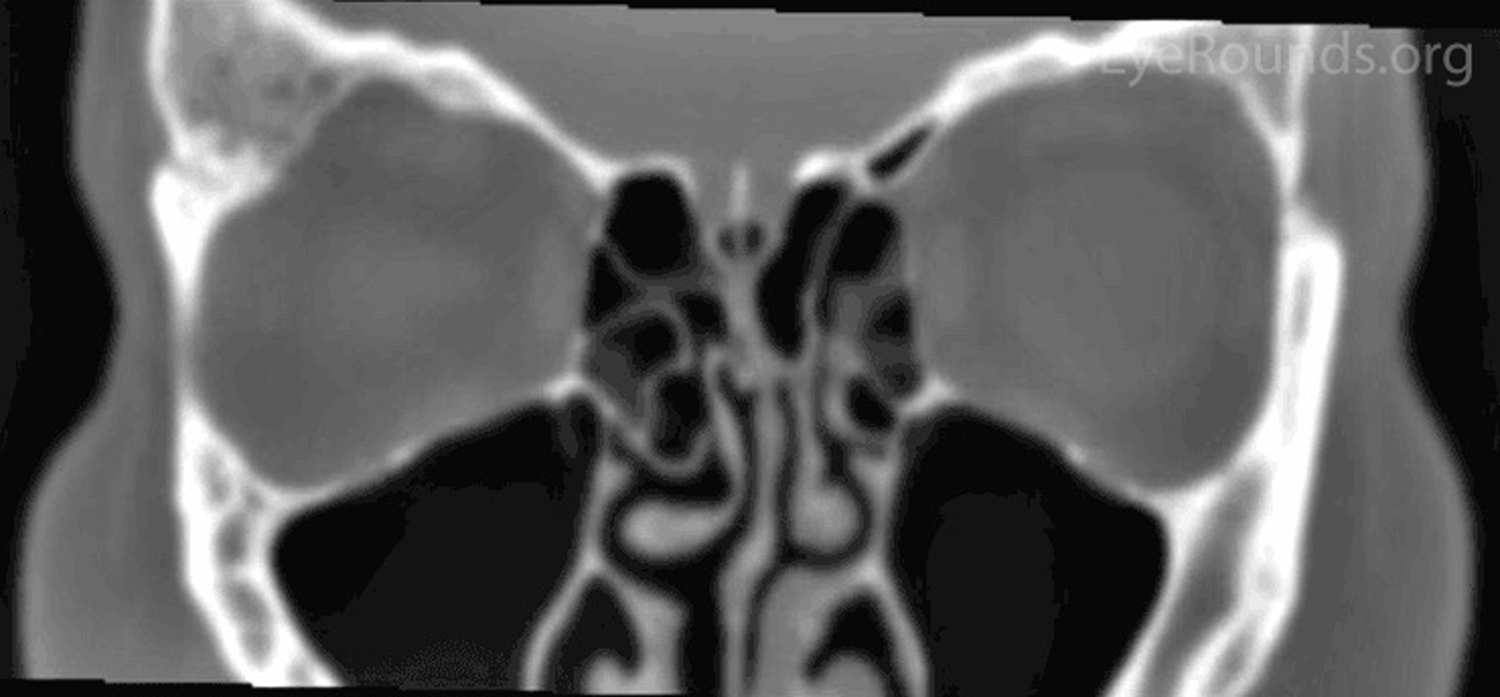
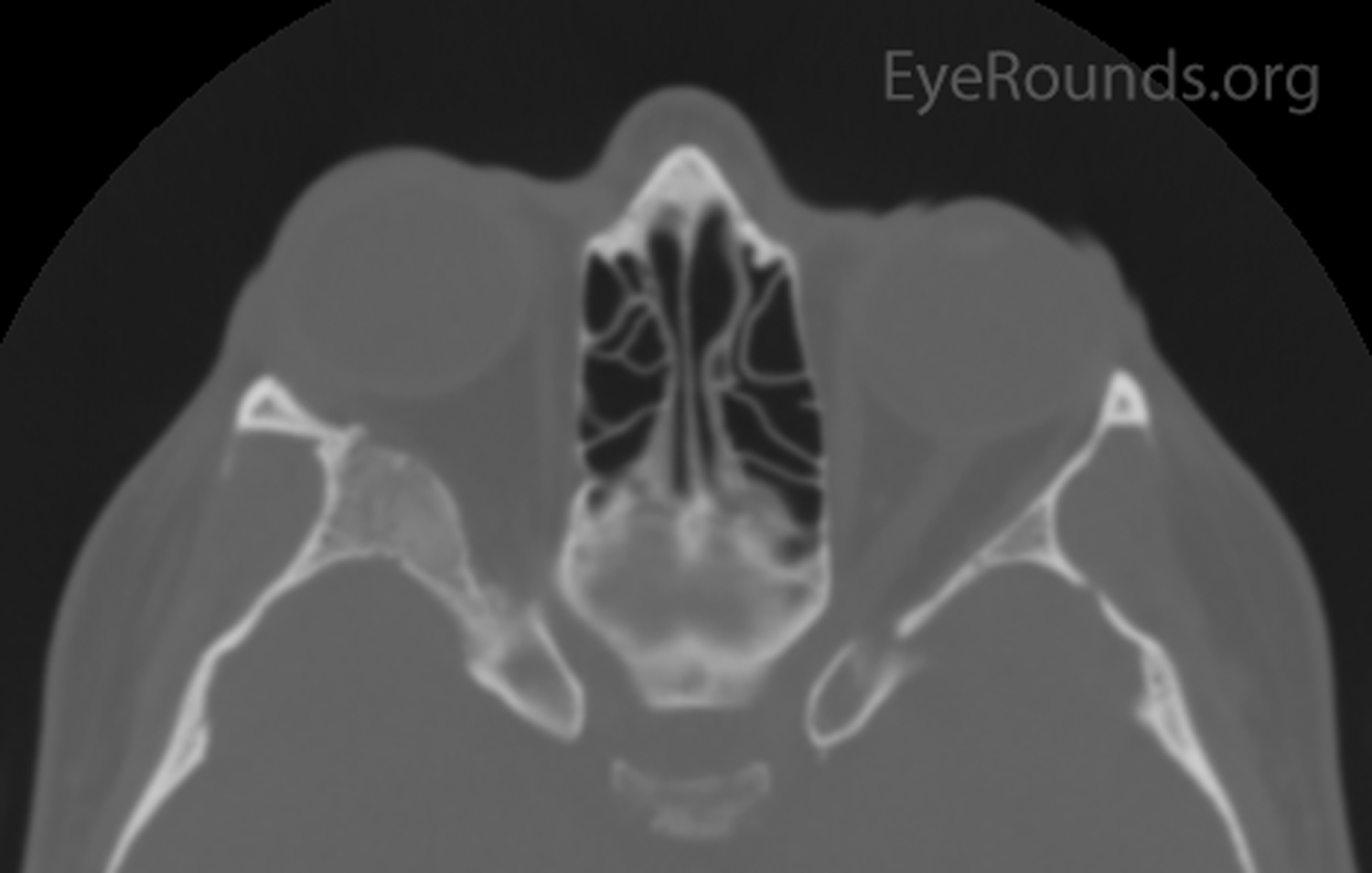
Figure 1: Orbital CT showing expansion of the right greater wing of the sphenoid bone with mottled appearance. (A: coronal view, B: axial view)
The patient's symptoms remained stable. However, her primary ophthalmologist noted 2 mm of increased proptosis over the course of 2 years, prompting a return to our clinic for repeat evaluation. She complained of intermittent blurry vision OU and increased proptosis for the last 6 months on the right side. Specifically, she reported that mascara had started to appear on her glasses, more on the right side than the left. She continued to deny pain with eye movement, diplopia, irritation, tenderness, or dry eye symptoms. During photography in clinic, she complained of binocular, horizontal diplopia in extreme right and left gazes.
| OD | OS | |
| External | proptotic | normal |
| MRD1 | 6 mm | 5 mm |
| MRD2 | 8 mm | 6 mm |
The patient had a CT of the orbits without contrast on the day of her clinic visit (Figure 2), in which medial displacement of the right lateral rectus muscle and a stable ground glass lesion of the right greater wing of the sphenoid bone were detected. There was no evidence of stenosis of the optic canal, and the globes were unremarkable. Based on her stable exam, the fibrous dysplasia was deemed unchanged. Her intermittent blurry vision was attributed to mild surface disease given her bilateral rapid tear break up time. Options were discussed, including observation, eyelid surgery to improve symmetry, medications such as bisphosphonates to improve pain, or surgery on the greater wing of the sphenoid. She elected for continued monitoring and would initiate artificial tears as needed.
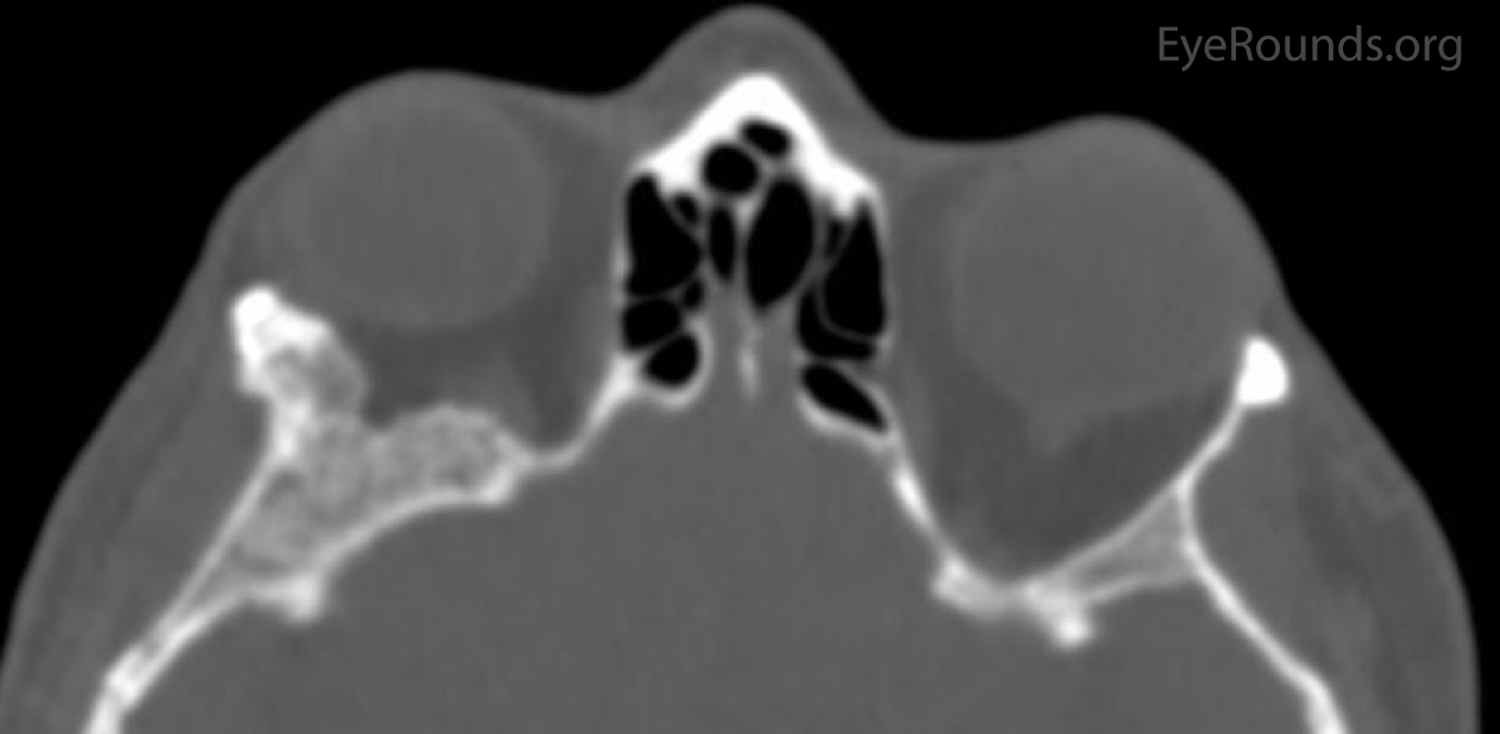
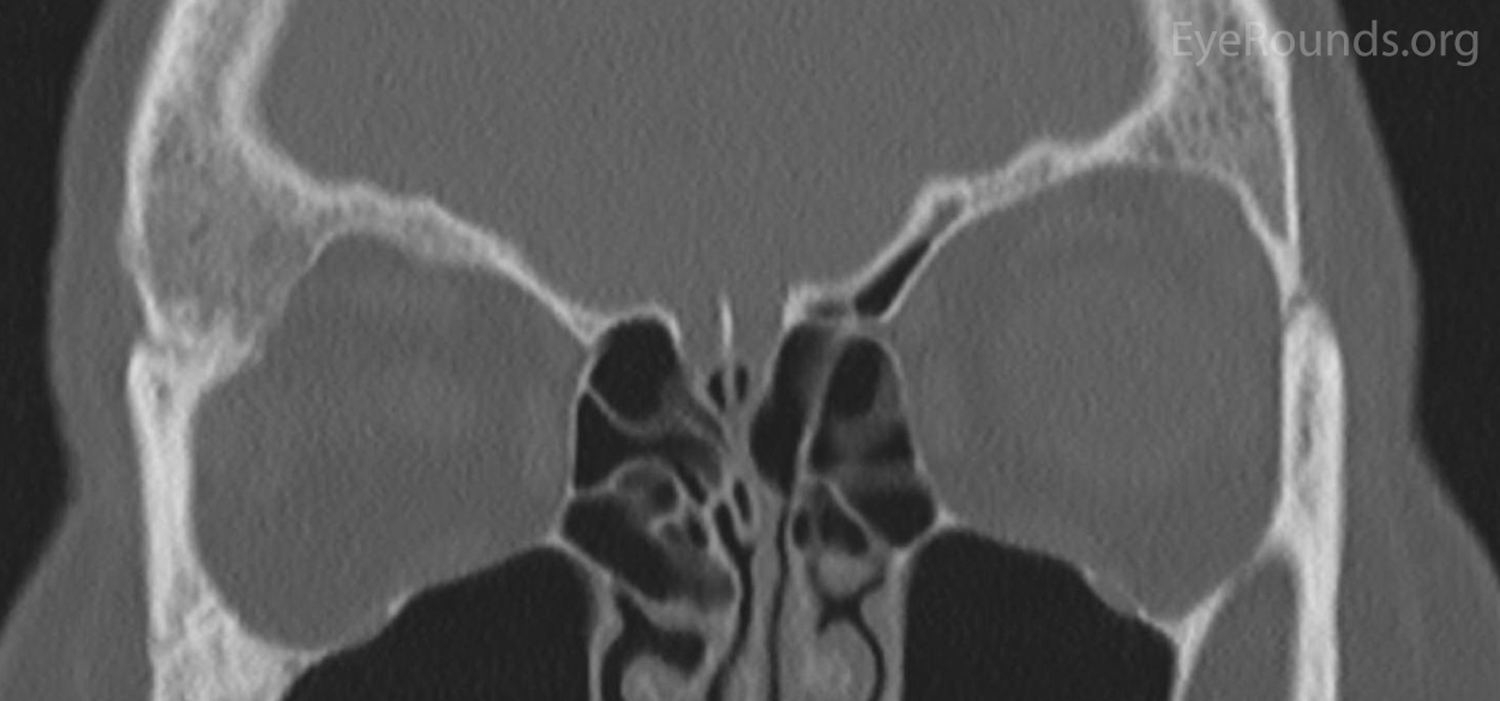
Figure 2: Orbital CT displaying a stable ground glass lesion of right greater wing of the sphenoid. (A: axial, B: coronal)
Craniofacial Fibrous Dysplasia
Fibrous dysplasia (FD) is a genetic, non-heritable, benign tumor of the bone, characterized by replacement of normal bone marrow by fibro-osseous tissue. Onset of symptoms typically occurs with diagnosis at 10 years of age on average.[1] FD affects males and females equally. There are three forms: monostotic (one site in the skeleton), polyostotic (multiple sites), and McCune Albright Syndrome (polyostotic + endocrine abnormalities). Monostotic FD accounts for 75% of all cases. The craniofacial bones are involved in approximately 20% of all FD cases.[2]
The cause of fibrous dysplasia stems from a missense mutation in the GNAS1 gene that codes for a stimulatory G protein. This mutated protein causes adenylyl cyclase activity to continue making cAMP, leading to the replacement of normal bone by immature stromal cells in a fibrous matrix. [1] Bone maturation slows down due to failure of osteoblastic/stromal cell differentiation, resulting in the formation of poorly organized bone.[2]
Patients with FD typically present with complaints of pain, swelling, asymmetry, or disfigurement. Specifically, primary complaints in patients with craniofacial involvement are headaches, proptosis, diplopia, and facial asymmetry. The most common neurological manifestations of FD are visual impairment and hearing loss. Visual impairment is estimated to occur in 20-80% of patients with craniofacial FD. Vision loss is typically chronic and secondary to a stenotic optic canal. Cases of acute vision loss are secondary to mucoceles or hemorrhages into pathologic bone tissue. [8] Sinus involvement and nasal obstruction can result in sinus collapse and epiphora.[3]
Craniofacial FD is a clinical diagnosis that relies on changes in the craniofacial exam and radiologic imaging. A thorough exam, including visual acuity, visual fields, and cranial nerve function, is vital in establishing a diagnosis of FD. Serum alkaline phosphatase is elevated in one third of FD patients and is occasionally drawn as part of the work up. Some physicians will monitor serum growth hormone (GH), as increased levels have been associated with more severe forms of craniofacial FD with optic neuropathy.[4]
Patients with suspected craniofacial FD should undergo a CT scan of the orbits without contrast to evaluate for characteristic lesions. CT helps to distinguish FD from a meningioma, which can share many clinical characteristics. The characteristic appearance of early FD is a cystic or sclerotic (ground glass) appearance in the frontal, maxillary, or sphenoid bone with smooth cortical margins. [4] As these lesions progress, they have more of a "pagetoid" pattern with alternating fibrous stroma and osseous change.[4] In the case of a sphenoid wing meningioma, the bone appears hyperostotic.
Encasement of the optic canal can be detected on a CT (50-90% of patients). Fortunately, optic nerve encasement does not have a strong correlation with deteriorating visual function and is not strongly associated with optic neuropathy in patients with FD.[5] This subject has been the matter of debate amongst ophthalmologists, who vary in the management of asymptomatic patients with radiologic evidence of optic nerve compression.
Approximately 20-30% of patients with FD have ocular involvement through a primary (osseous expansion) or a secondary process (aneurysmal bone cyst, mucocele).[5] The progression of the ocular symptoms from FD is gradual and insidious due to the slow growth of the bony lesion. While lesions are typically benign and slow-growing, knowing when to continue periodic follow-up and when surgical intervention is necessary is essential in managing patients with craniofacial FD. One must keep in mind that 1-2% of craniofacial FD patients will have malignant transformation.[6]
The treatment of FD is dependent on the disease severity, patient symptoms, and enlargement on imaging. Patients that are asymptomatic, have non-progressive FD, or who present only with mild-moderate proptosis/discomfort are typically observed with yearly imaging to carefully monitor for growth of the lesion. Patients with active FD (rapid growth, neurologic changes on exam) require imaging and immediate referral for possible surgical evaluation.
Medical management of FD has been more recently studied. Some studies link bisphosphonates (pamidronate) with improvement in pain in FD patients. [7] Steroids are a short-term option for patients with acute visual loss, but there is no evidence of steroid use as a long-term agent for preservation of vision.
Patients with acute or chronic vision loss and evidence of FD on imaging typically undergo therapeutic optic nerve decompression. The superior approach for resection of dysplastic bone for optic nerve decompression involves a frontotemporal extradural approach. [8] This provides the best access for orbital bone resection and subsequent reconstruction, most commonly of the sphenoid, ethmoid, and frontal bones. [8] Recurrent FD following optic nerve decompression is uncommon, and the extent of decompression is a large determinant of possible recurrence due to the increased chance of residual bone containing FD. [8]
As previously mentioned, management of asymptomatic patients with radiological evidence of optic nerve encasement is a topic of debate. A recent meta-analysis supports a conservative approach of observation and periodic imaging, as opposed to prophylactic optic nerve decompression. [9] The conservative approach is driven by the demonstration that in most patients with FD, optic nerve encasement does not progress with age, and thus there is no increased risk of developing an optic neuropathy. [9] Additionally, there is a risk of iatrogenic visual loss when performing a prophylactic decompression.
Craniofacial FD warrants a multidisciplinary approach to ensure that visual impairment caused by FD is identified and treated properly to improve visual outcomes. Periodic follow-up with ophthalmologic evaluation, visual field testing, and imaging is important to monitor craniofacial FD, whether active or quiescent.
EPIDEMIOLOGY AND ETIOLOGY
|
SIGNS
|
SYMPTOMS
|
TREATMENT/MANAGEMENT
|
Smith CA, De Andrade LM, Carter KD, Shriver EM. Fibrous Dysplasia. EyeRounds.org. posted October 12, 2017; Available from https://eyerounds.org/cases/260-fibrous-dysplasia.htm

Ophthalmic Atlas Images by EyeRounds.org, The University of Iowa are licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License.

Address
University of IowaLegal
Related Links
