
INITIAL PRESENTATION
Chief Complaint: Acute-onset diplopia and bilateral ophthalmoplegia
History of Present Illness:
A 47-year-old man with a history of Crohn’s disease presented to an outside emergency room reporting sudden-onset of diplopia. A month prior to this, he had diarrhea for 3 weeks, which was presumed to be due to a flare of his Crohn’s disease. He was treated with oral prednisone (40 mg daily), which he took for three days. On the fourth day, he woke up with binocular, oblique diplopia and was unable to move his eyes in any direction.
Thinking the diplopia was related to starting the prednisone, he stopped taking it. He was evaluated in the ER by Neurology, who noted complete bilateral ophthalmoplegia. CT head, MRI brain with contrast, and MRA head with contrast were obtained and were all unremarkable. Acetylcholine receptor antibody testing was obtained due to concern for myasthenia gravis and was also negative. He was started on doxycycline to empirically treat for possible Lyme disease.
The patient was referred to the Neuro-Ophthalmology service at the University of Iowa. At that time, in addition to persistent diplopia and ophthalmoplegia, he complained of reduced color vision and headaches that he attributed to eye strain. He also reported eye pain with upward gaze.
Past Ocular History: None
Medical History:
Medications:
Allergies: No known Allergies
Family History: : The patient’s father has multiple sclerosis with speech and mobility limitations. No family ocular problems.
Social History: Former smoker, quit 13 years ago. No illicit drugs, no alcohol.
Review of Systems: The patient denied any variability of symptoms throughout the day, extremity weakness, sensory disturbance, or pain elsewhere. He denied difficulties with swallowing, speech, or breathing. No rashes, joint pain, or known insect bites.
OCULAR EXAMINATION
Differential Diagnosis:
CLINICAL COURSE
Laboratory workup performed prior to presentation at the University of Iowa Neuro-Ophthalmology clinic included negative acetylcholine receptor antibodies (AChR), muscle-specific kinase antibodies, TSH and thyroid peroxidase antibodies, syphilis and Lyme serologies, ANCA, ANA, SSA/B, and creatine kinase. Chest CT was negative for thymoma, thymic hyperplasia, or lymphadenopathy. Electromyography (EMG) was also normal.
Review of the previous head imaging (CT, MRI, MRA) showed no evidence of brainstem stroke, cavernous sinus lesion, or enlargement of extraocular muscles to suggest myositis or thyroid eye disease.
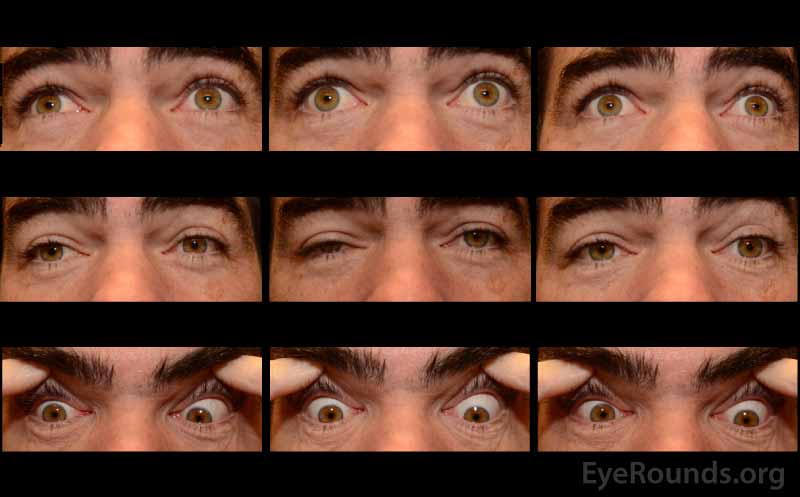
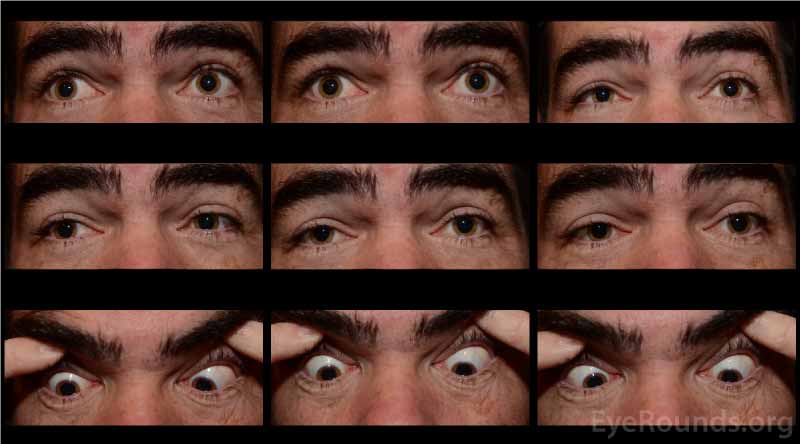
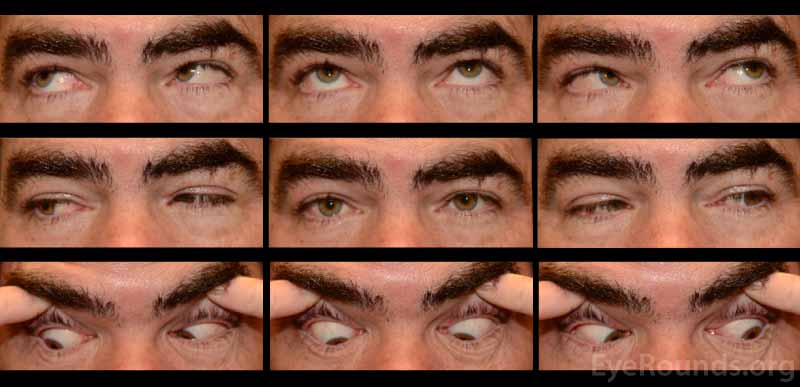
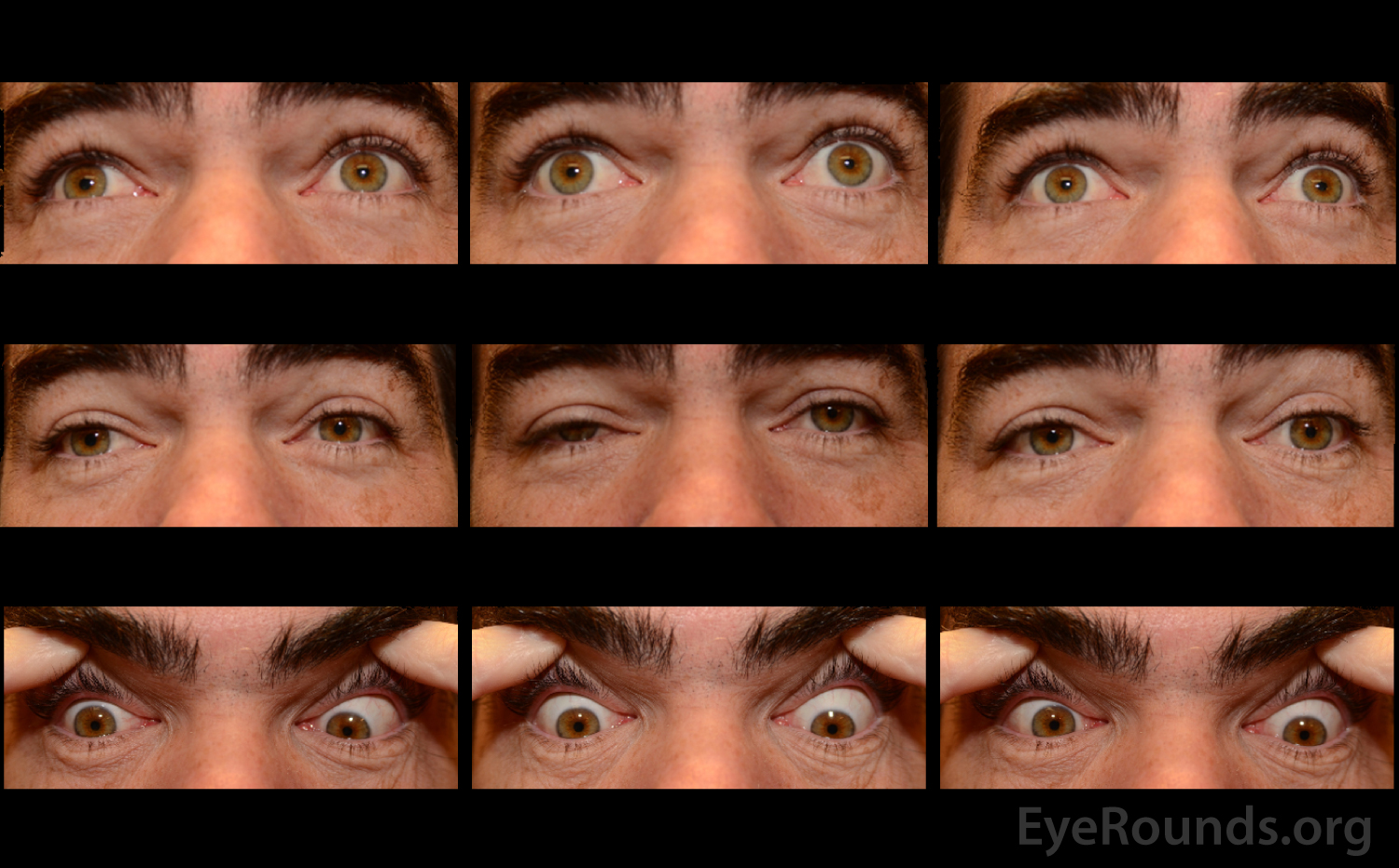
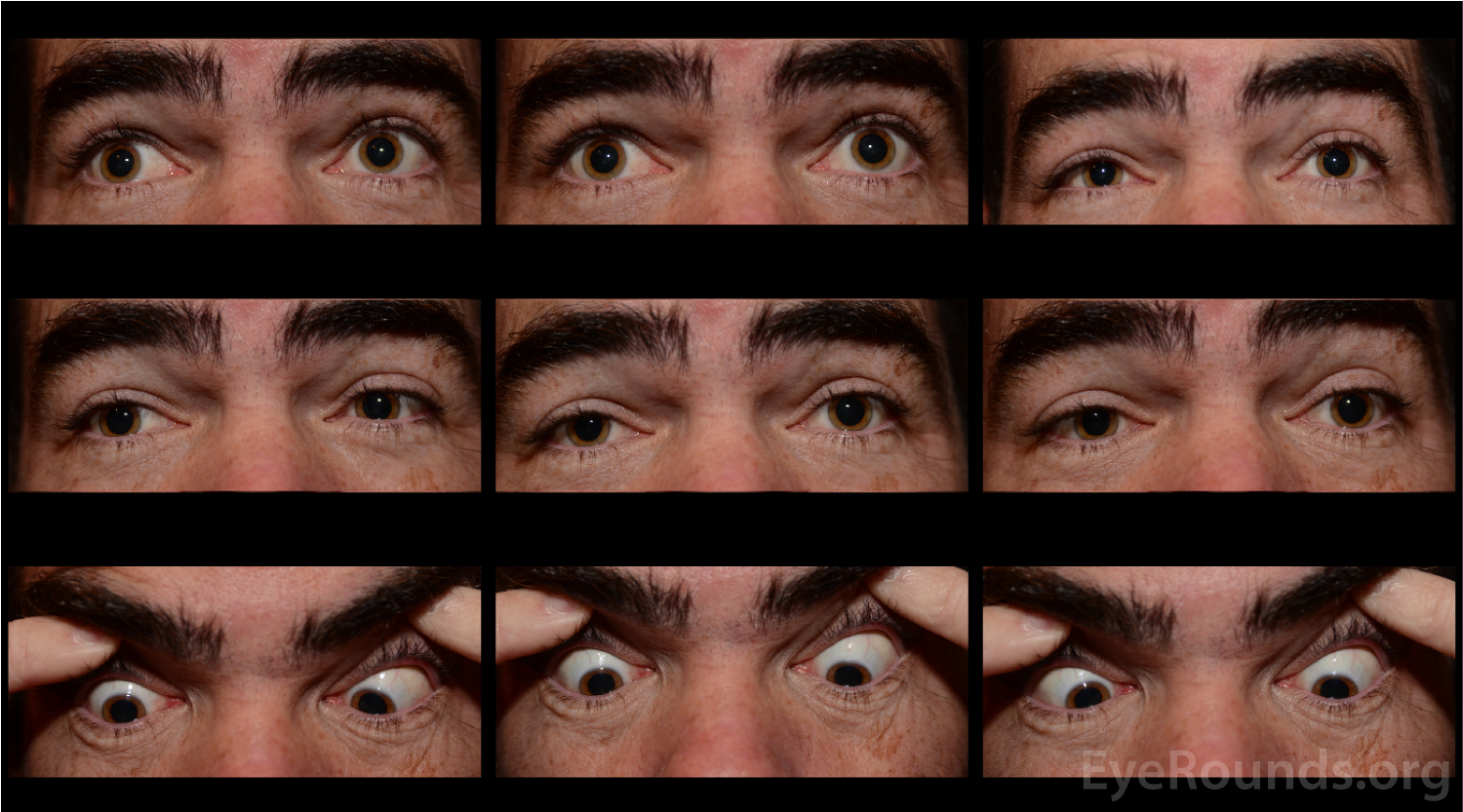
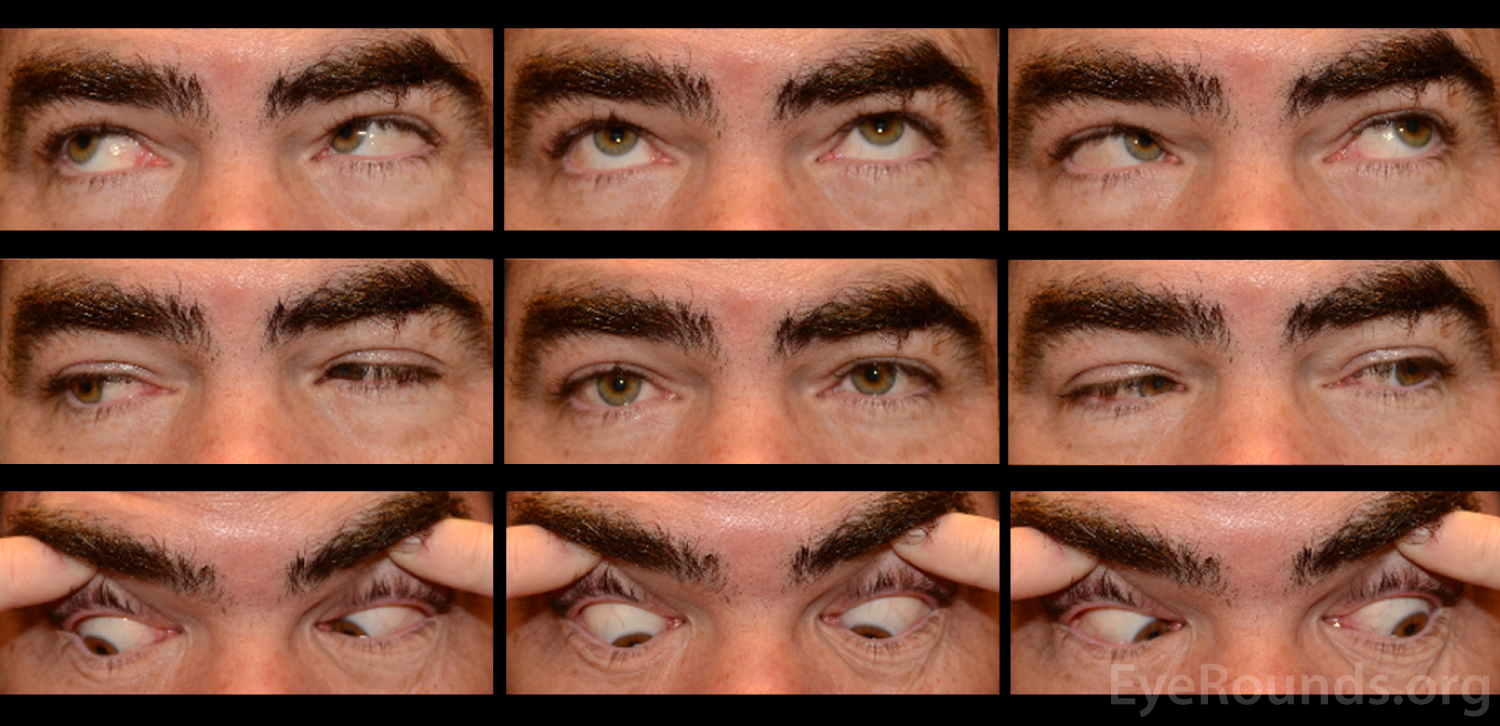
A neostigmine test was performed in clinic and the patient was felt to have moderate improvement in his ptosis and ophthalmoplegia OU (Figures 1 and 2). Given the positive neostigmine test, seronegative ocular myasthenia was initially thought to be the likely diagnosis. The patient was restarted on an augmentation of oral prednisone (started at 10mg, increased to 20mg over four weeks) as well as oral pyridostigmine (60mg three times daily). He was encouraged to wear an eye patch alternating between eyes to relieve the double vision. Repeat AChR antibody testing was ordered. Despite absence of areflexia and ataxia, GQ1b antibody testing was also obtained to rule out Miller Fisher Syndrome (MFS). The AChR antibody test returned negative a second time, but the GQ1b antibody test returned positive.
The patient called three weeks later to report that his diplopia seemed to have worsened, but his eye motility was improving. He had stopped taking the pyridostigmine because of side-effects, but he continued to take prednisone 20mg daily. He did not feel that either medication had helped his symptoms.
Given the lack of response to pyridostigmine and prednisone, absence of characteristic fatigability associated with myasthenia gravis, and negative AChR antibody testing, ocular myasthenia was felt to be less likely. Despite the lack of ataxia and areflexia, MFS was felt to be more likely given his positive GQ1b antibody and history of diarrheal illness weeks before the onset of ocular symptoms. The slow improvement in eye motility over time seemed consistent with the self-resolving nature of MFS. The patient was therefore tapered off the prednisone.
The patient returned for follow up two months later, at which time he complained of continued diplopia. On exam, his ptosis had completely resolved (Figure 3). His ductions were full OU, although he continued to have a large esotropia (25 prism diopters in primary position) and left hypertropia (5 prism diopters in primary position). He was encouraged to continue wearing an eye patch. Given his progress and the characteristically good prognosis of MFS, it was felt that he would make a full spontaneous recovery with time.
DIAGNOSIS: Miller Fisher Syndrome
DISCUSSION
EPIDEMIOLOGY
Miller Fisher Syndrome (MFS) is a rare subtype of Guillain-Barre Syndrome (GBS), an immune-mediated polyneuropathy characterized by acute ascending motor weakness and areflexia [1,2]. MFS was first described by James Collier in 1932, but the syndrome is named after C. Miller Fisher who, in 1956, proposed that MFS was a subtype of GBS since both show an elevated cerebrospinal fluid (CSF) protein concentration [3]. The incidence of MFS is not established, but the global incidence of GBS is approximately 1-2 per 100,000. Various studies estimate the incidence of MFS to be between 1-7% of GBS incidence in the West and 15-25% in Asia. MFS affects all age groups (although adults are more frequently affected than children), and shows a slight male predominance [4,5].
PATHOPHYSIOLOGY
As with GBS, most patients with MFS describe symptoms of either an upper respiratory tract or gastrointestinal infection, with upper respiratory tract infection being more common [4,5] prior to the onset of neurological symptoms. The infection can occur anytime from 3 days to 6 weeks before the onset of neurological symptoms, with a median time of 8 days [5,6]. In most cases, the causative organism is not identified. However, when it is, Campylobacter jejuni is the leading cause followed by Haemophilus influenzae. Cytomegalovirus, Mycoplasma pneumoniae, Epstein-Barr virus, Group A streptococcus, Coxiella burnetii, Pasturella multocida, Helicobacter pylori, Aspergillus, and varicella zoster have been more rarely identified preceding the onset of MFS [5].
The most sensitive finding for MFS is the presence of GQ1b antibodies [4,5]. These antibodies are present in more than 85% of patients with MFS, and antibody levels correlate with the clinical severity of the disease (particularly the ophthalmoplegia) [5]. The GQ1b ganglioside is a component of the neuronal cell membrane and has areas of identical structure to lipo-oligosaccharides found on the membranes of certain strains of Campylobacter and Haemophilus [4,5].Therefore, through the mechanism of “molecular mimicry,” GQ1b antibodies that develop in response to the infectious agent cross-react with the gangliosides on cranial nerves 3, 4, and 6, muscle spindle afferent fibers, cerebellar granule cells, and dorsal root ganglia, thereby giving rise to the ophthalmoplegia, ataxia, and areflexia [4,5]. The degeneration of neuronal tissue in these areas is mediated through complement attack at the neuromuscular junction [5].
SIGNS AND SYMPTOMS
The classic triad of clinical features in MFS is ophthalmoplegia, ataxia, and areflexia [3,4]. The most common presenting symptom is diplopia, followed by ataxia [4,5].
Ophthalmoplegia is the most common sign in MFS, especially in the acute phase of the illness. In most cases, the ophthalmoplegia is external, bilateral, and symmetric, but cases of unilateral or internal ophthalmoplegia (affecting the pupillary reflexes rather than the extraocular muscles) have been reported [5]. The ataxia in MFS can be severe enough that the patient loses his or her ability to walk independently and, similar to the ophthalmoplegia, the clinical severity correlates with levels of GQ1b antibody [5]. Although our patient’s normal reflexes were a factor that initially steered us away from a diagnosis of MFS, areflexia is the least common presenting symptom of the triad and the least specific for the syndrome [4].
Other less frequent signs and symptoms of MFS include dysesthesias, ptosis, facial weakness, limb weakness, dysphagia, headache, optic neuritis, photophobia, blurred vision, and periocular pain. MFS can present with features that overlap with GBS and other GBS subtypes, on occasion progressing to quadriparesis and respiratory failure requiring mechanical ventilation. In patients with MFS without features of overlap, 1% require assisted ventilation. However, patients with MFS-GBS overlap require mechanical ventilation even more often than patients with GBS alone [5].
Limited forms of MFS have been reported, in which the clinical triad of ophthalmoplegia, ataxia, and areflexia is not fully present. Isolated acute ophthalmoplegia (AO) associated with GQ1b antibodies is a limited form of MFS [4]. This subtype of MFS best fits the clinical picture of our patient who had neither ataxia nor areflexia. Incomplete forms of MFS that show only isolated ptosis, ataxia, or mydriasis have also been reported [6].
Due to the overlap in GBS subtypes and the existence of limited forms of subtypes, it is increasingly hypothesized that the syndromes under the GBS umbrella form a continuous spectrum rather than distinct entities. The presence of GQ1b antibodies in multiple subtypes of GBS seems to support this theory [5]. In addition to having a positive GQ1b antibody, our patient also had a positive GM1 antibody, which is a ganglioside antibody associated with a subtype of GBS called acute motor axonal neuropathy (AMAN) [4].
DIFFERENTIAL DIAGNOSIS
When considering the differential diagnosis of MFS, myasthenia gravis (MG) is important to include. These two conditions overlap both clinically and physiologically. Like MFS, MG can present with acute complete bilateral external ophthalmoplegia, ptosis, diplopia, orbicularis oculi weakness, bulbar weakness, and a Cogan lid twitch (a brief over-elevation of the upper lids when gaze is shifted from downward to straight ahead) [7]. Fluctuation and fatigability (weakness exacerbated by exertion), though sometimes subtle, are clinical features that would suggest MG over MFS. In MG, weakness can either be localized, most commonly to the extraocular muscles, or generalized, typically involving the proximal extremity muscles in addition to the extraocular and bulbar muscles. Weakness in the small muscles of the hand can be a specific sign for MG, and weakness that is chronic or recurrent is also suggestive of MG [8], although a small proportion of patients with MFS can have recurrent attacks [9]. Tendon reflexes and sensation are normal in MG. Presence of a thymoma, although only found in about 10% of patients with MG, would decrease suspicion for MFS [8].
MFS and MG overlap in pathophysiology at the neuromuscular junction. IgG from the serum of patients with MFS demonstrates direct binding and blockade of nicotinic acetylcholine receptor channels, similar to MG [10, 11]. IgG in MFS also appears to have blocking effects at the presynaptic terminal, masking any decremental response that would be seen at the postsynaptic terminal in electrophysiological tests [10,11]. This may explain why MFS does not present with the same fluctuating and fatigable weakness of MG. Complement activity in MFS also appears to act at the presynaptic terminal [7].
The edrophonium test is considered to be a reliable diagnostic test for MG, though it is much more sensitive than specific [12, 13]. A positive edrophonium test has a positive likelihood ratio for MG of 15, while a negative test has a negative likelihood ratio of 0.11 [14]. A literature review found that positive edrophonium tests have been reported in cases of Lambert-Eaton myasthenic syndrome, botulism, GBS, and amyotrophic lateral sclerosis in addition to congenital and drug-induced myasthenic syndromes [12]. Yonemoto et al. were the first to report a case of MFS with a false-positive edrophonium test [15].
Co-occurrence of MG and MFS is rare, but has been documented in the literature. Yuan et al. reviewed four reported cases in which patients were diagnosed with both MG and MFS, and described a fifth [16]. In three of the cases, there was an established diagnosis of MG years before the patient presented with MFS. In another case, the diagnosis of MFS was made before the diagnosis of MG, and in another, the two conditions were diagnosed at the same time. Three of the five cases presented with a positive edrophonium test and four with the presence of AChR antibodies [16].
TESTS, IMAGING, LAB WORKUP
MFS is a diagnosis that can often be made clinically and supported by the presence of elevated CSF protein concentration and GQ1b antibodies. GQ1b antibodies are usually present within 1 week of the onset of symptoms [5]. The vast majority of neuroimaging studies are normal. Neuroimaging is mostly used to exclude other etiologies for the patient’s presentation, although there can be abnormalities like faint enhancement or increased T2 signal in the brainstem, cerebellum, posterior columns, and nerve roots in rare cases [4,5]. Autopsies have also demonstrated brainstem lesions and peripheral nerve demyelination in rare instances [5]. EMG and nerve conduction studies, which can be quite useful in diagnosing other subtypes of GBS, are often normal in MFS [4]. The most consistent EMG finding in MFS is the absence of a soleus H-reflex, a sign of proximal conduction impairment affecting only group Ia neurons. Conduction abnormalities in Ia spinocerebellar afferent fibers of muscles spindles may explain the ataxia and areflexia [17-20]. A much less consistent sign is abnormal f-wave latency, a sign of conduction impairment in the spinal roots, limb girdle plexuses, and proximal segments of nerves, likely due to demyelination [18].
TREATMENT
MFS is a self-limiting condition and has a very good prognosis, especially the limited forms like AO [4]. Almost all patients completely recover by 6 months regardless of treatment [5,21]. Recovery time does not appear to be affected by age, sex, clinical severity at peak, or time between onset and peak symptoms [5]. There have been no randomized controlled trials to evaluate treatments for MFS, but retrospective studies have shown that intravenous immune globulin (IVIG) hastens recovery while having no impact on long-term outcome [4,5]. There is no evidence that either plasmapheresis or corticosteroids have any impact on the onset of recovery or outcome [1,4,5]. Regardless, different combinations of IVIG, plasmapheresis, and corticosteroids have been used to treat patients with MFS. At the time our patient was diagnosed with MFS, enough time had elapsed from the onset of his symptoms that IVIG would likely not have been of great benefit to him.
EPIDEMIOLOGY
|
SIGNS & SYMPTOMS
|
ETIOLOGY
|
TREATMENT/MANAGEMENT
|
References
Wang C, Wilson C, Thurtell M. Cavernous Hemangioma: Miller Fisher Syndrome: 47-year-old male with sudden onset diplopia and bilateral ophthalmoplegia EyeRounds.org. Posted Feb. 10, 2022. Available from http://www.EyeRounds.org/cases/300-miller-fisher-syndrome.htm.

Ophthalmic Atlas Images by EyeRounds.org, The University of Iowa are licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License.

Address
University of IowaLegal
Related Links

{kind=link}
{kind=link}
{kind=link}