
INITIAL PRESENTATION
Chief Complaint: Six months of persistently blurry vision in the left eye
History of Present Illness:
A 68-year-old Caucasian man was referred to the University of Iowa for six months of declining vision in his left eye. There was no associated pain, flashes, floaters, or diplopia. Vision in the right eye was unaffected. At the initial onset of symptoms, he presented to his local ophthalmologist, who performed a YAG capsulotomy bilaterally. Vision continued to be “blurry,” and on repeat exam, the patient was noted to have vitreous cell in the left eye. An Ozurdex implant was injected into the left eye, which yielded a temporary improvement in vision. However, after 10 weeks, the patient’s vision began to decrease again with recurrence of apparent vitritis. Diagnostic vitrectomy showed a monoclonal B cell population. At this point, the patient was referred to UIHC for further evaluation and treatment.
Past Ocular History:
Past Medical History:
Medications:
Allergies: No known drug allergies
Family History: : No family history of cancer or ocular disease
Social History:
Review of Systems:
OCULAR EXAMINATION
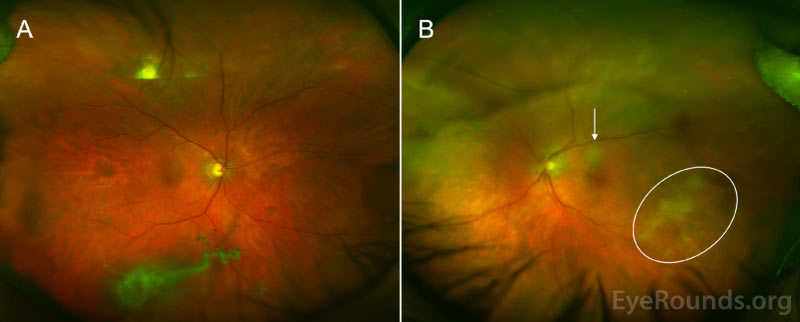
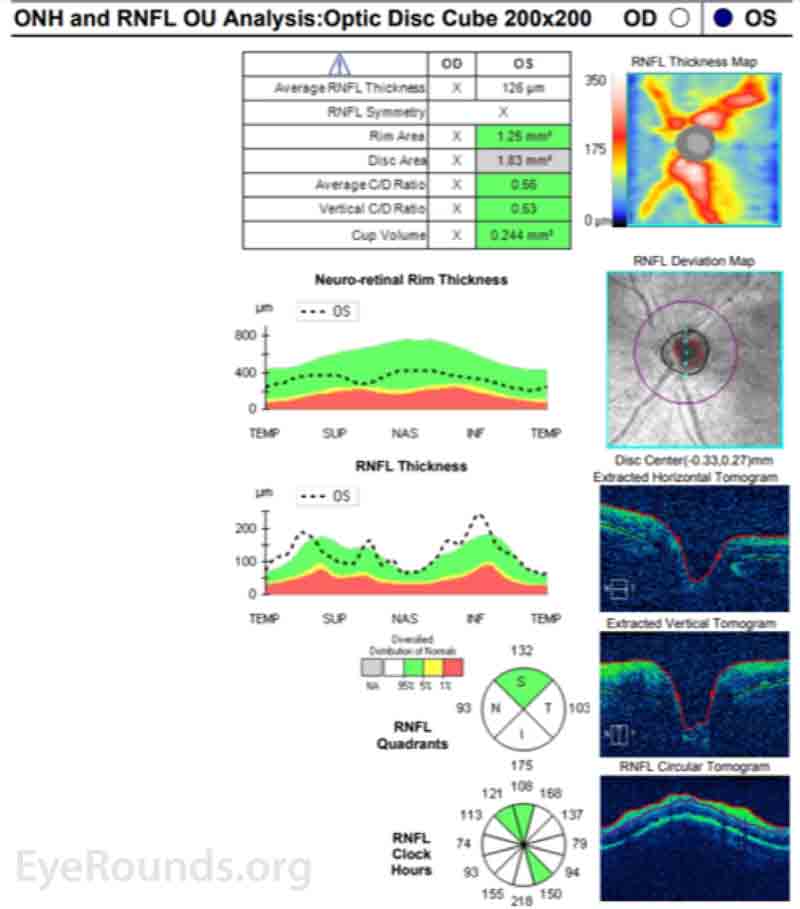
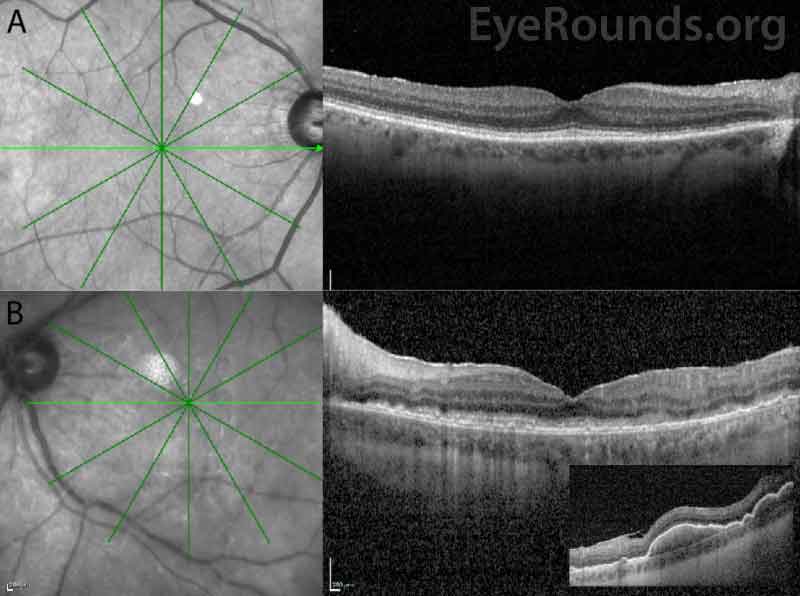
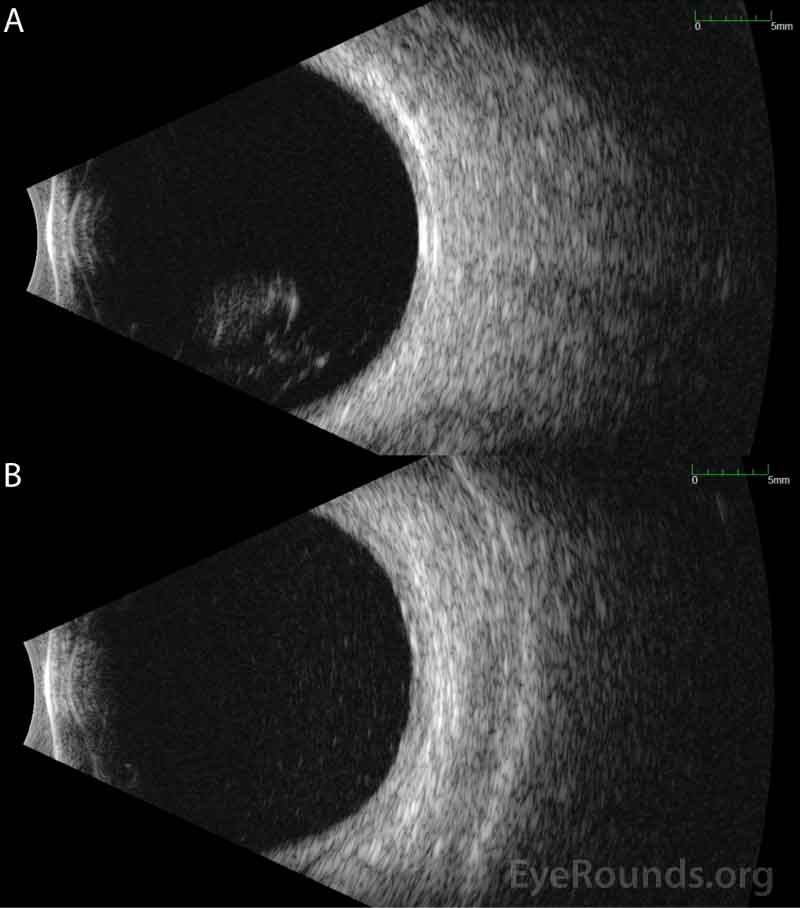
Differential Diagnosis:
CLINICAL COURSE
This patient had a clinical picture consistent with primary vitreoretinal lymphoma of the left eye. To rule out central nervous system involvement, the patient received an urgent oncology evaluation and workup, including MRI of the brain, PET scan, and lumbar puncture. Fortunately, there was no evidence of systemic or CNS disease.
Although the patient reported no visual changes in the right eye, the presence of vitreous cells and a large central vitreous opacity OD raised concern for early involvement of the right eye as well. The patient underwent diagnostic pars plana vitrectomy of the right eye; pathology revealed low cellularity with fibrinous material and no identifiable lymphoma cells. Given the absence of subretinal infiltrates and lymphoma cells, the right eye was observed closely without clear indication for treatment.
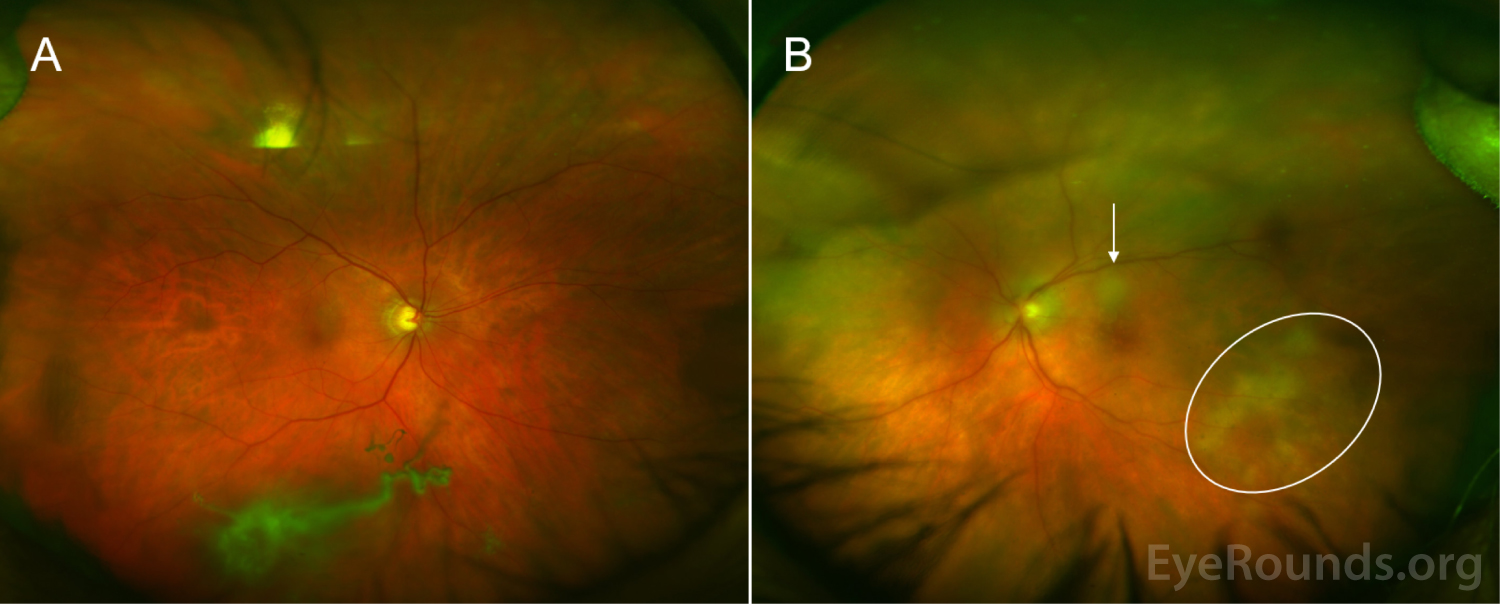
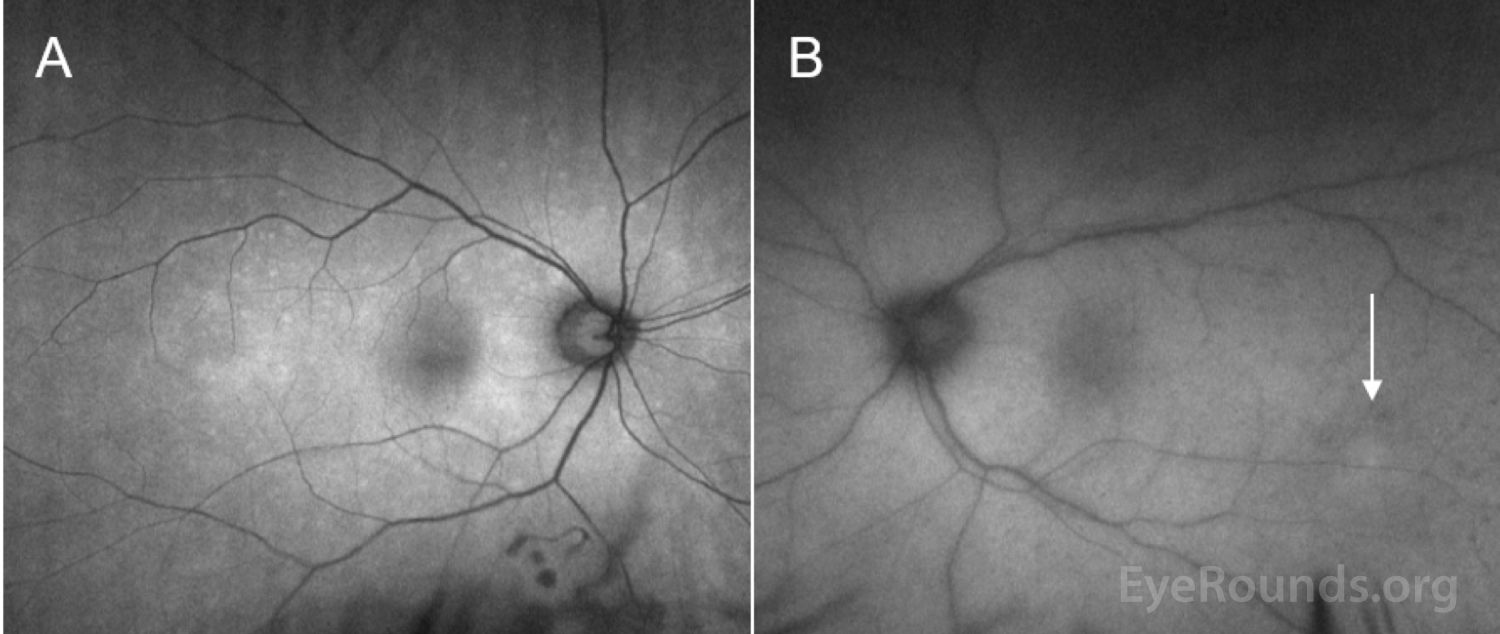
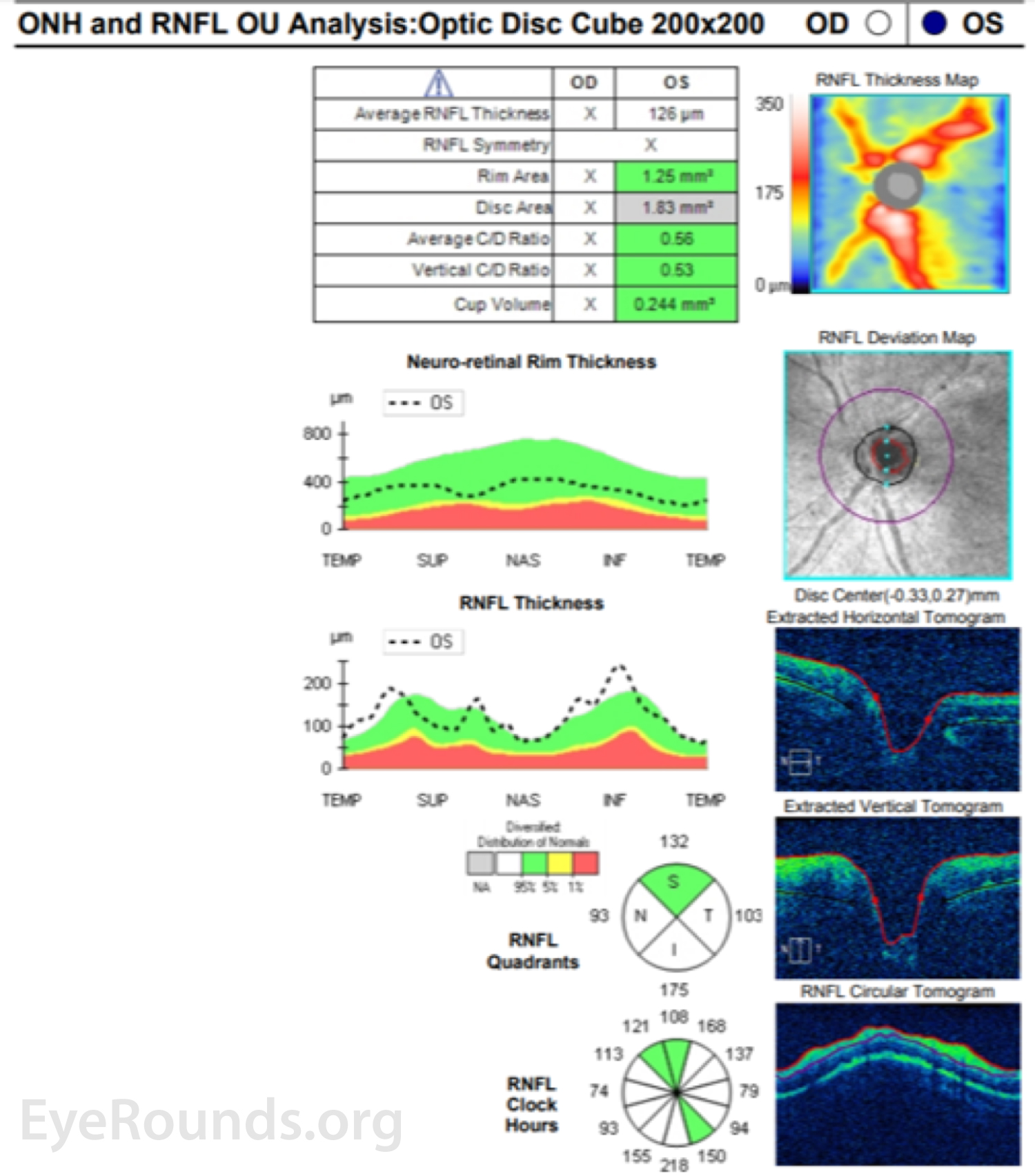
The patient initially opted for systemic chemotherapy (methotrexate and rituximab) combined with intraocular methotrexate, which he tolerated well. After two cycles of chemotherapy, the patient declined further systemic treatment and continued with intraocular therapy alone. He received 400 micrograms intravitreal methotrexate in the left eye twice weekly for one month, then weekly for one month, then monthly thereafter. The left eye responded well to the methotrexate, with marked improvement of disc edema and subretinal infiltrates with chorioretinal atrophy after one month of therapy (Figure 6).
Throughout the course of treatment, vision gradually improved to 20/30 OS and remained stable without signs of lymphoma recurrence at nine months of follow up at the time of publication of this article. He continues to receive monthly methotrexate injections OS and surveillance brain imaging at 3 month intervals.
DIAGNOSIS: Primary vitreoretinal lymphoma
DISCUSSION
This 68-year-old man with no significant oncologic or ophthalmic history presented with six months of progressive visual decline and was found to have persistent vitritis refractory to steroids, subretinal deposits, potential optic disc infiltration, and a monoclonal B-cell population on diagnostic vitrectomy. This clinical picture was consistent with primary vitreoretinal lymphoma (PVRL) of the left eye. The patient did not have any signs of central nervous system (CNS) involvement, and ultimately opted for local treatment with intravitreal methotrexate injections.
Etiology / Pathophysiology:
PVRL is a very rare, high-grade malignancy of B-cell origin with a poor prognosis. Originating from the vitreous or retina, this malignancy is classified as a primary intraocular lymphoma (PIOL). Among the intraocular lymphomas, primary vitreoretinal lymphoma is the most common. However, PVRL still comprises less than 1% of all intraocular tumors, and is estimated to affect fewer than 2000 patients in the United States annually [1, 2]. Although it is difficult to quantify the true incidence of this disease due to its rarity, cases have been increasing in the past 25 years [3]. The mean age of diagnosis is 60 years of age, with increased rates occuring in those who are immunocompromised or immunosuppressed [4, 5].
PVRL is considered a manifestation or subtype of primary CNS lymphoma (PCNSL). At this time, the pathogenesis of PCNSL is poorly understood, and it is unclear whether CNS and ocular manifestations occur independently or whether the originating primary lymphoma subsequently spreads to other sites. Regardless of the mechanism, ocular lymphoma is intricately associated with the CNS, and PVRL often precedes or occurs concurrently with CNS lymphoma. Up to 90% of patients with PVRL eventually develop CNS involvement, which may present in the brain, spinal cord, or meninges [4]. Conversely, a primary CNS lymphoma can precede vitreoretinal disease –15-25% of patients with primary CNS lymphoma go on to develop ocular dissemination of disease, which is an independent risk factor for lower survival [6]. Due to the propensity for CNS involvement, PVRL has a high mortality rate, with a median overall survival of 58 months [7].
Signs/Symptoms:
The clinical features of PVRL are quite heterogenous, and signs and symptoms can be extremely varied between patients. Disease is bilateral in approximately 80% of cases [4, 8]. The most common ophthalmic complaints include blurred or hazy vision, painless vision loss, and floaters [5]. Less commonly, patients report photophobia, red eyes, or ocular pain [9]. If lymphoma has spread to the brain, it often grows extremely rapidly and can cause neurological symptoms, including cognitive dysfunction, behavior changes, hemiparesis, ataxia, and seizures [4, 9].
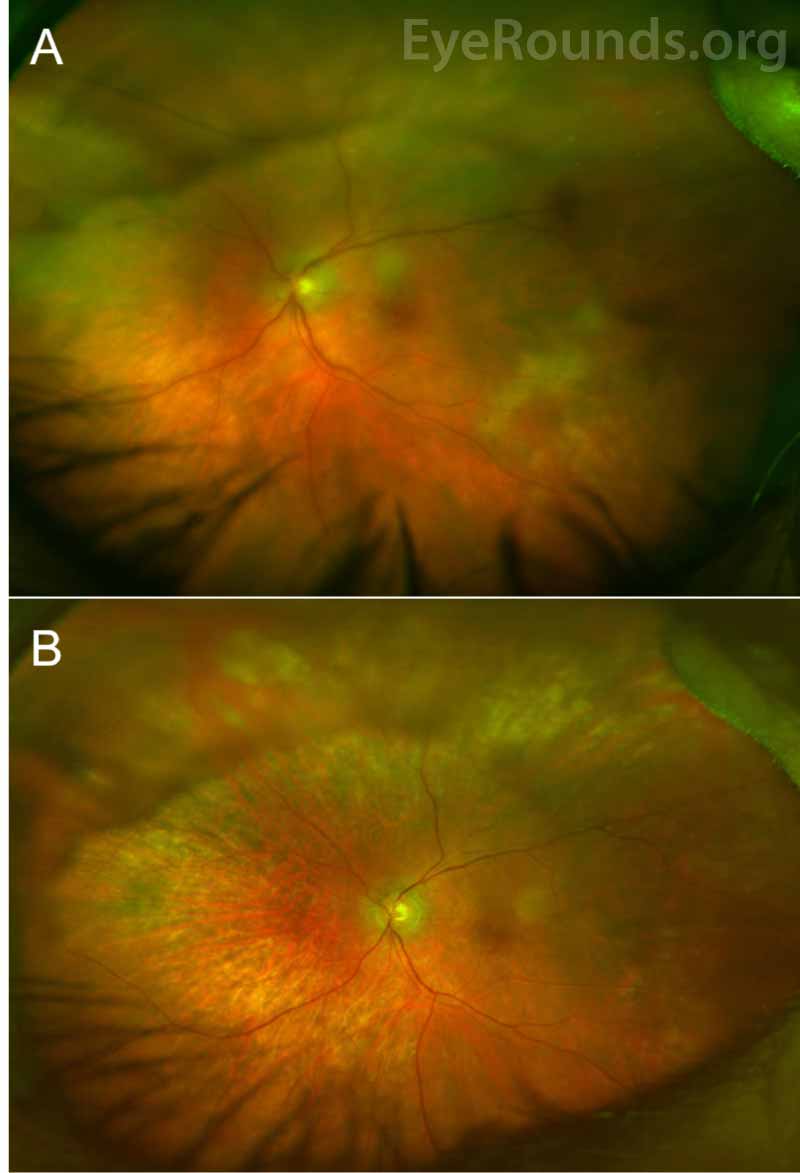
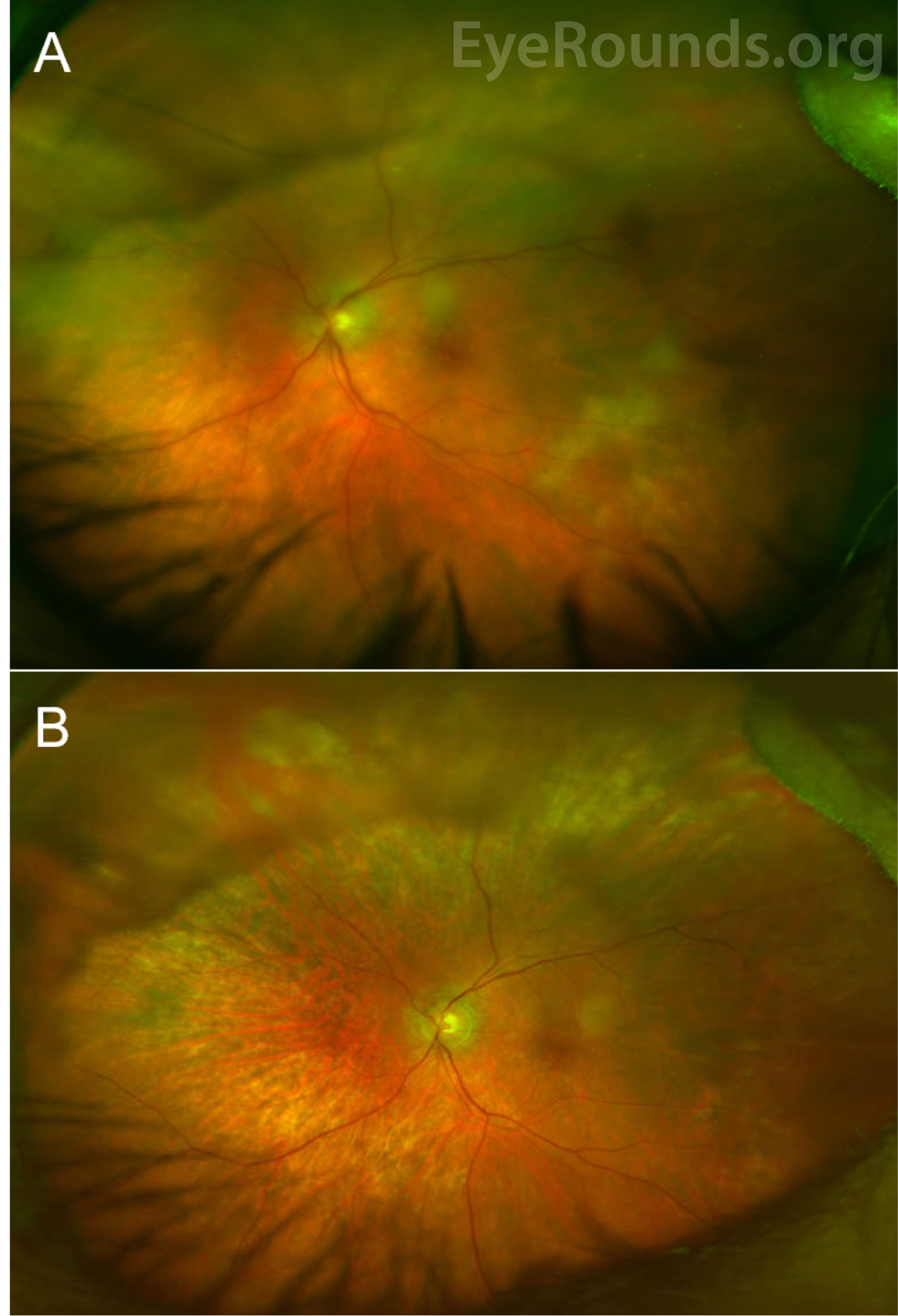
Ocular examination reveals vitritis in the majority of cases, which appears as homogenous sheets, strands, and clumps of vitreous cells and haze. However, visual acuity is often much better than expected for the extent of vitritis. PVRL can masquerade as posterior uveitis for months to years prior to diagnosis, and it is not uncommon for patients to have a history of multiple courses of steroids for their presumed chronic or recurrent uveitis. The nonspecific presentation often delays diagnosis, with literature reporting the mean time of symptom onset to diagnosis as 21 - 40 months [8, 10]. As such, a high index of suspicion is necessary to identify PVRL in a timely manner, especially in an elderly patient with a history of uveitis refractory to standard steroid treatment.
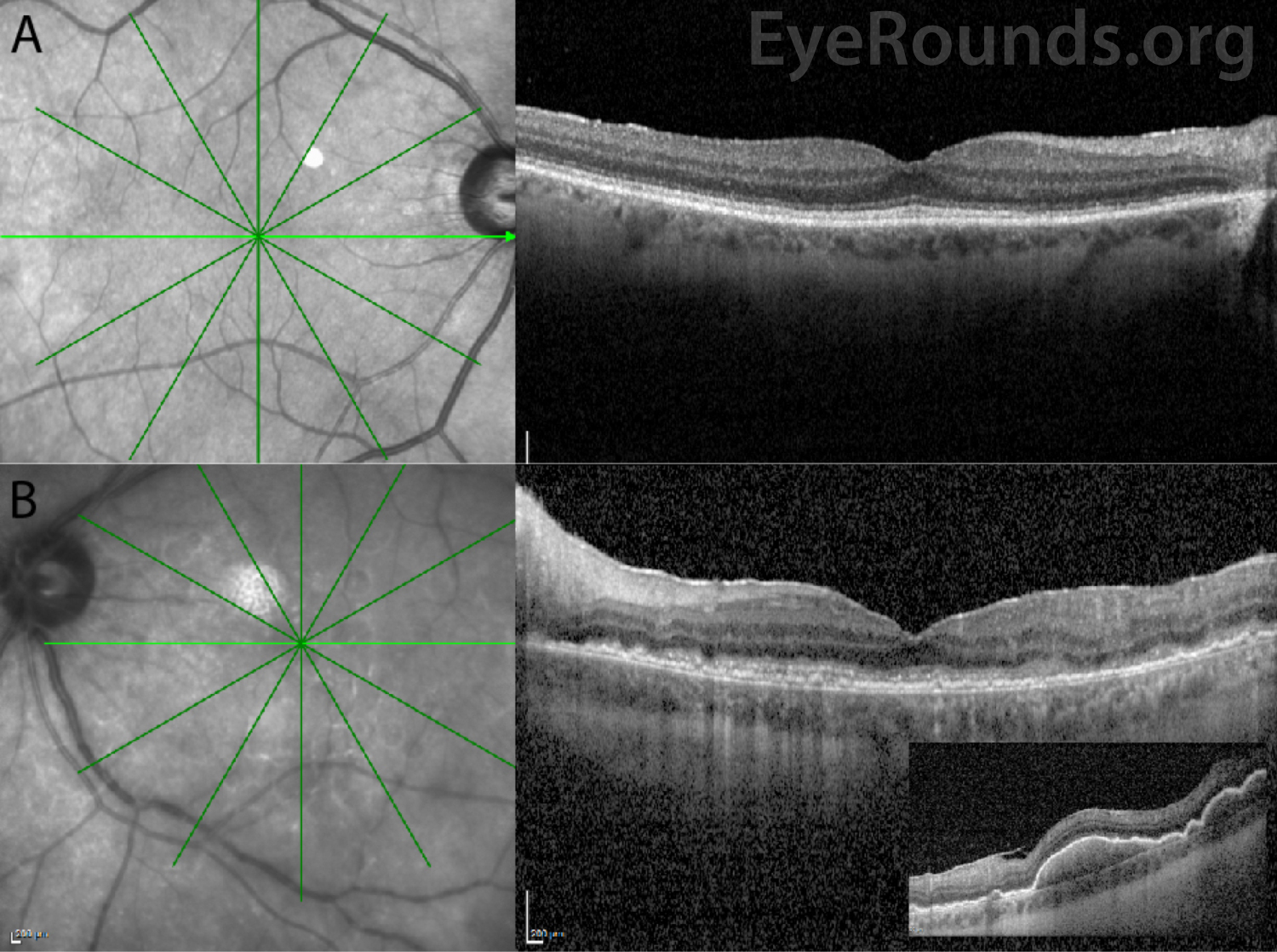
Vitreous cells can infiltrate the retinal pigment epithelium (RPE) to form lesions, and the presence of multiple flat, creamy, orange-yellow subretinal masses on funduscopic exam is considered by some to be pathognomonic of PVRL [4, 5, 11]. However, it is important to know that these lesions are not present in all patients with PVRL, and many patients present with a completely normal dilated fundus exam. Others may have multifocal or confluent lesions associated with solid irregular RPE detachments or RPE atrophy with subretinal fibrosis. Cystoid macular edema – a common finding in other forms of uveitis or with administration of intravitreal methotrexate – is rarely seen in PVRL.
Anterior segment findings are less common, and the eye is usually white and quiet. If abnormalities are noted, they can include anterior cell or flare, keratitic precipitates, pseudohypohyon, and – rarely – iris and angle infiltration [9].
Testing and Imaging Work-up:
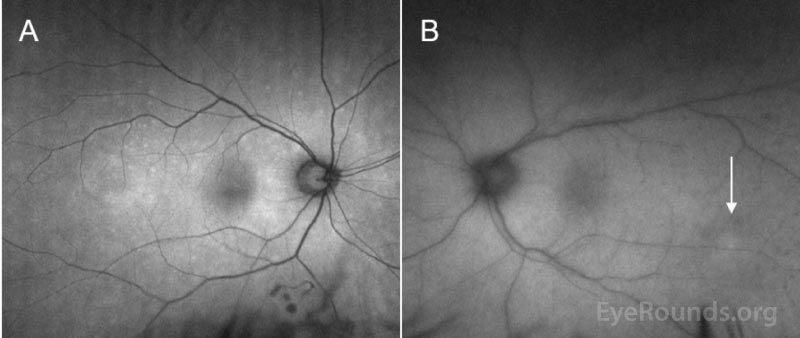
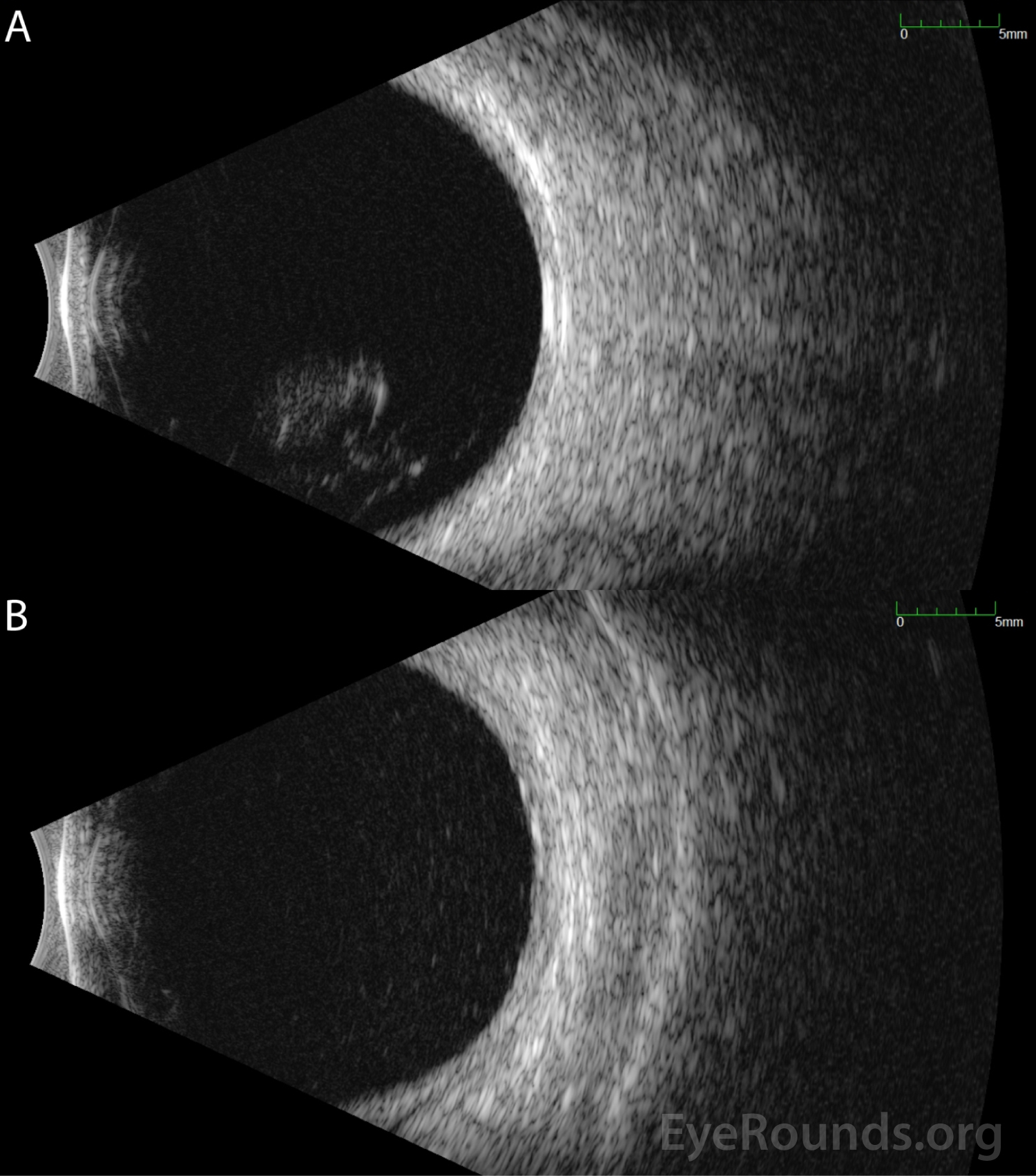
Ocular imaging can be a useful ancillary tool in establishing the diagnosis of PVRL, and is often the first step that is pursued prior to invasive biopsy procedures. Fundus autofluorescence may reveal either hyperautofluorescent or hypoautofluorescent deposits [12]. Fluorescein angiography can also show hypofluorescent or hyperfluorescent changes at the level of the RPE such as granularity, blockage, and late staining [3, 13]. OCT is useful for finding and characterizing retinal deposits and subretinal infiltrates. The most common finding on OCT in PVRL is a collection of hyperreflective nodular lesions in the RPE [3]. Finally, B-scan ultrasound imaging can show sheets of vitreous debris, retinal detachment, elevated chorioretinal lesions, and optic nerve widening, although none of these changes are specific for PVRL [5].
The gold standard for diagnosis is intraocular cytological examination, which is obtained by vitrectomy or retinochoroidal biopsy. Biopsy may be supplemented by immunohistochemistry, flow cytometry, and polymerase chain reaction analysis for immunoglobulin gene rearrangements. Although a variety of molecular studies are available (identifying biomarkers such as CD20+ or monoclonal kappa or lambda B-cell populations), one that is highly specific and sensitive is the IL-10:IL-6 ratio. Because the proliferation of B lymphocytes in the vitreous is thought to be incited by the growth factor IL-10, an IL-10:IL-6 ratio > 1 in aqueous or vitreous humor is highly suggestive of, but not diagnostic for, PVRL [1, 4, 12]. Due to the small sample volume of intraocular biopsies, as well as the high rate of false positives and false negatives, repeat biopsy may be necessary in ~25% of cases to definitively establish the correct diagnosis [1]. Recently, screening for a mutation in MYD88, a protein associated with the innate immune system, in vitreous and aqueous humor samples has also shown promise in serving as a diagnostic tool for the detection of PVRL [14, 15].
Once a diagnosis of PVRL has been made, it is critical to pursue a complete systemic work-up and imaging to stage the disease. A full ocular exam should be performed bilaterally, even if the patient has no ophthalmic complaints. It is also important to rule out a primary systemic lymphoma, which includes a CT scan of the chest and abdomen as well as a testicular ultrasound in elderly males. PVRL involvement of the CNS can be assessed by neuroimaging and lumbar puncture. A brain MRI showing a single homogenously enhancing periventricular lesion is suggestive of lymphoma spread to the brain, and is best differentiated from other CNS tumors with diffusion-weighted imaging [4]. If results are inconclusive or the clinical picture is suspicious, a stereotactic biopsy of the brain lesion may be pursued. Lumbar puncture with CSF analysis is most useful for assessing leptomeningeal involvement [9].
Treatment / Management / Guidelines:
It is imperative to use a multi-team approach in the management of PVRL as collaboration between the ophthalmologist, oncologist / hematologist, cytopathologist, and neurologist will optimize the patient’s treatment regimen.
The first step in management of PVRL is to assess for systemic and CNS involvement of disease (as above). If CNS involvement is evident, the treatment approach will involve local treatment of the intraocular lymphoma plus systemic chemotherapy with high-dose methotrexate (HD-MTX) based regimens, and sometimes whole-brain radiation [4]. .
If the disease appears to be localized solely to the eye, then therapy consists of local ocular treatments with or without the option of systemic therapy. Conservative regimens to treat PVRL focus solely on eradication of lymphoma cells in the eye using intravitreal injections of methotrexate and/or rituximab [1, 2]. Ocular irradiation has also been used, but has fallen out of favor in recent years due to its undesirable adverse effects [2]. More aggressive treatment regimens are aimed not only at achieving local control, but also preventing CNS dissemination of disease, even if clinical signs of CNS disease are not yet present. As such, a wide variety of systemic chemotherapy regimens have been used in conjunction with intraocular therapy [16]. Specific regimens vary widely and are largely dependent on the institution.
A complete understanding of the best strategies for management of PVRL is still being established, and there is currently no strong consensus for treatment approach due to the rare nature of the disease and lack of randomized clinical trials. Current data do not support one approach over another, and clinical outcomes have been shown to be similar regardless of the use of ocular or systemic treatment regimens [7]. Continued research is required to further understand which treatments portend the best response, lower recurrence rates, and optimize visual outcomes.
EPIDEMIOLOGY
|
SIGNS
|
SYMPTOMS
|
TREATMENT/MANAGEMENT
|
References
Yom KH, Fortenback CR, Binkely EM. Primary Vitreoretinal Lymphoma (PVRL): 68-year-old man with six months of persistently blurry vision in the left eye. EyeRounds.org. Posted September 17, 2020. Available from http://www.EyeRounds.org/cases/303-Primary_Vitreoretinal_Lymphoma_(PVRL).htm.

Ophthalmic Atlas Images by EyeRounds.org, The University of Iowa are licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License.

Address
University of IowaLegal
Related Links

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}