
INITIAL PRESENTATION
Chief Complaint: Establish care, family history of retinal detachment
History of Present Illness:
A 10-year-old boy presented to establish care due to a strong family history of retinal detachment (eight individuals across three generations). At the age of three, he was seen by a pediatric ophthalmologist who noted an optically empty vitreous and diagnosed him with presumed Wagner syndrome (hereditary vitreoretinopathy).
At his presenting visit, he had no visual complaints. He had a history of moderate myopia, but no hearing loss, no arthritis, and no ocular symptoms.
Past Ocular History: Myopia (about -5 diopters spherical equivalent)
Medical History:
Medications:
Allergies: None
Family History: :
Social History: Patient's family just moved to Iowa City
Review of Systems: Negative except for what is detailed in the history of present illness
OCULAR EXAMINATION
| OD | OS | |
|---|---|---|
| Lids/lashes | Normal | Normal |
| Conjunctiva/sclera | Clear and Quiet | Clear and Quiet |
| Cornea | Clear | Clear |
| Anterior chamber | Deep and quiet | Deep and quiet |
| Iris | Normal architecture | Normal architecture |
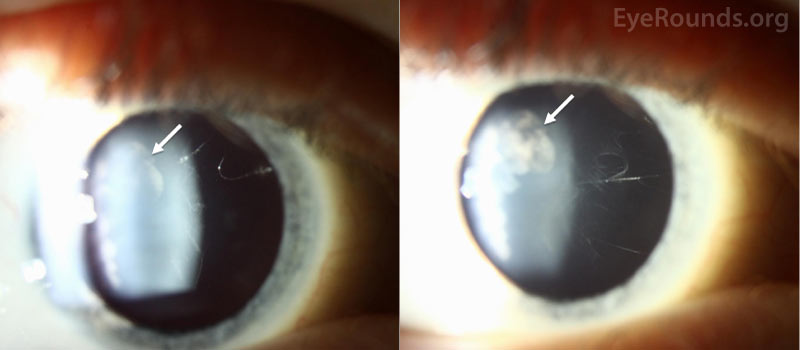
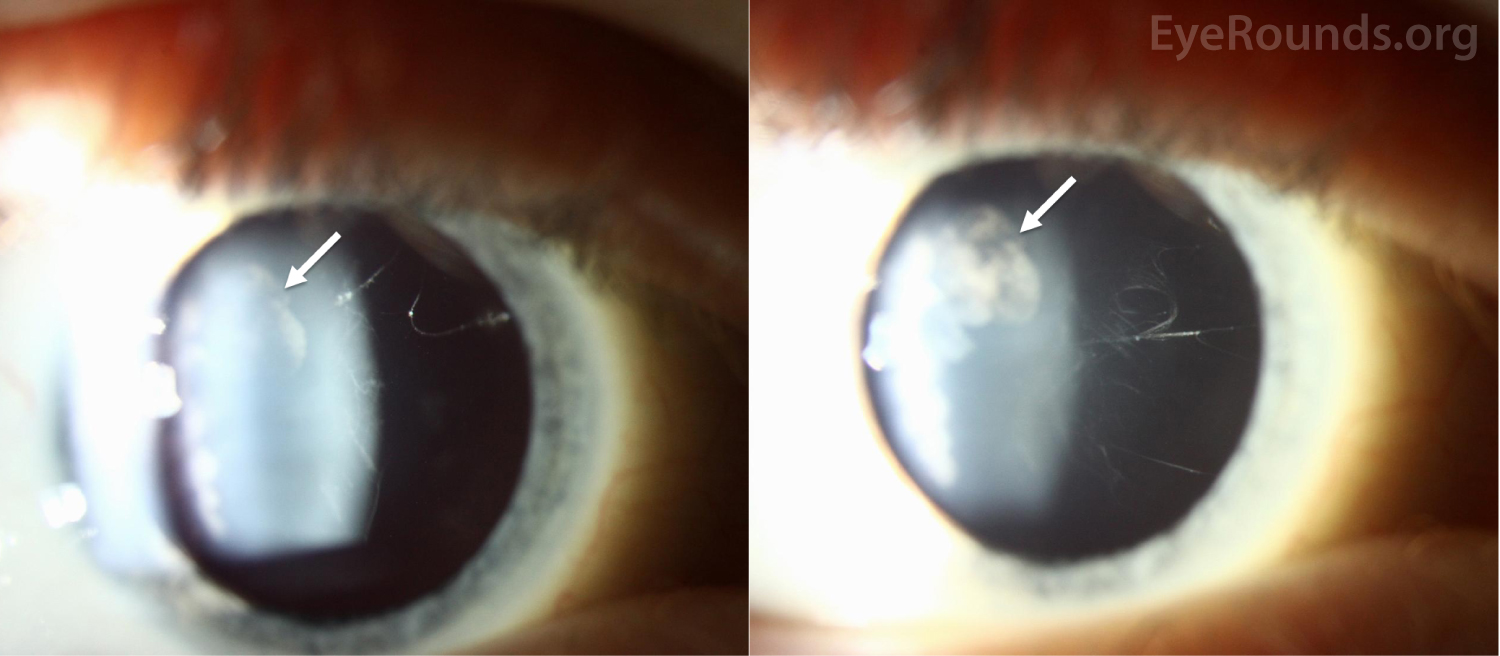
| Lens | Trace to 1+ cortical cataract temporally | Trace to 1+ cortical cataract temporally |
| Vitreous | Optically empty with vitreous veils | Optically empty with vitreous veils |
| OD | OS | |
|---|---|---|
| Disc | Normal | Normal |
| Cup-to-disc ratio | 0.3 | 0.3 |
| Macula | Sharp foveal light reflex | Sharp foveal light reflex |
| Vessels | Normal | Normal |
| Periphery | Lattice degeneration inferiorly | Lattice degeneration inferiorly |
CLINICAL COURSE
Molecular screening for causes of a heritable vitreoretinopathy was recommended, and a blood sample was collected. Results revealed a pathologic variant in COL2A1, consistent with Type I Stickler syndrome.
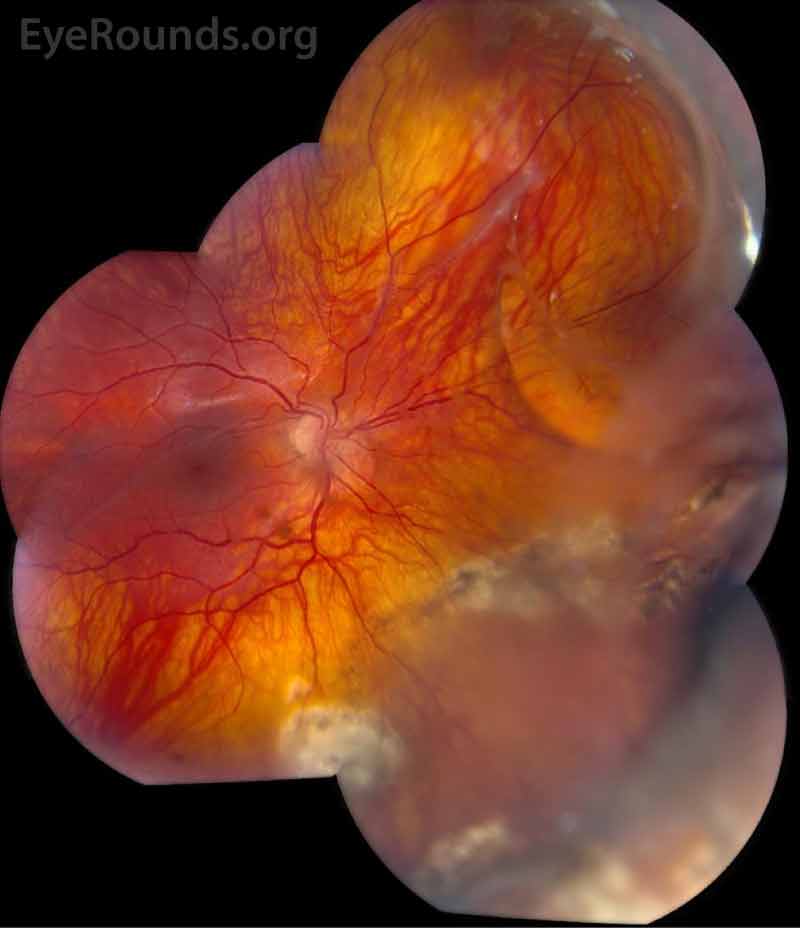
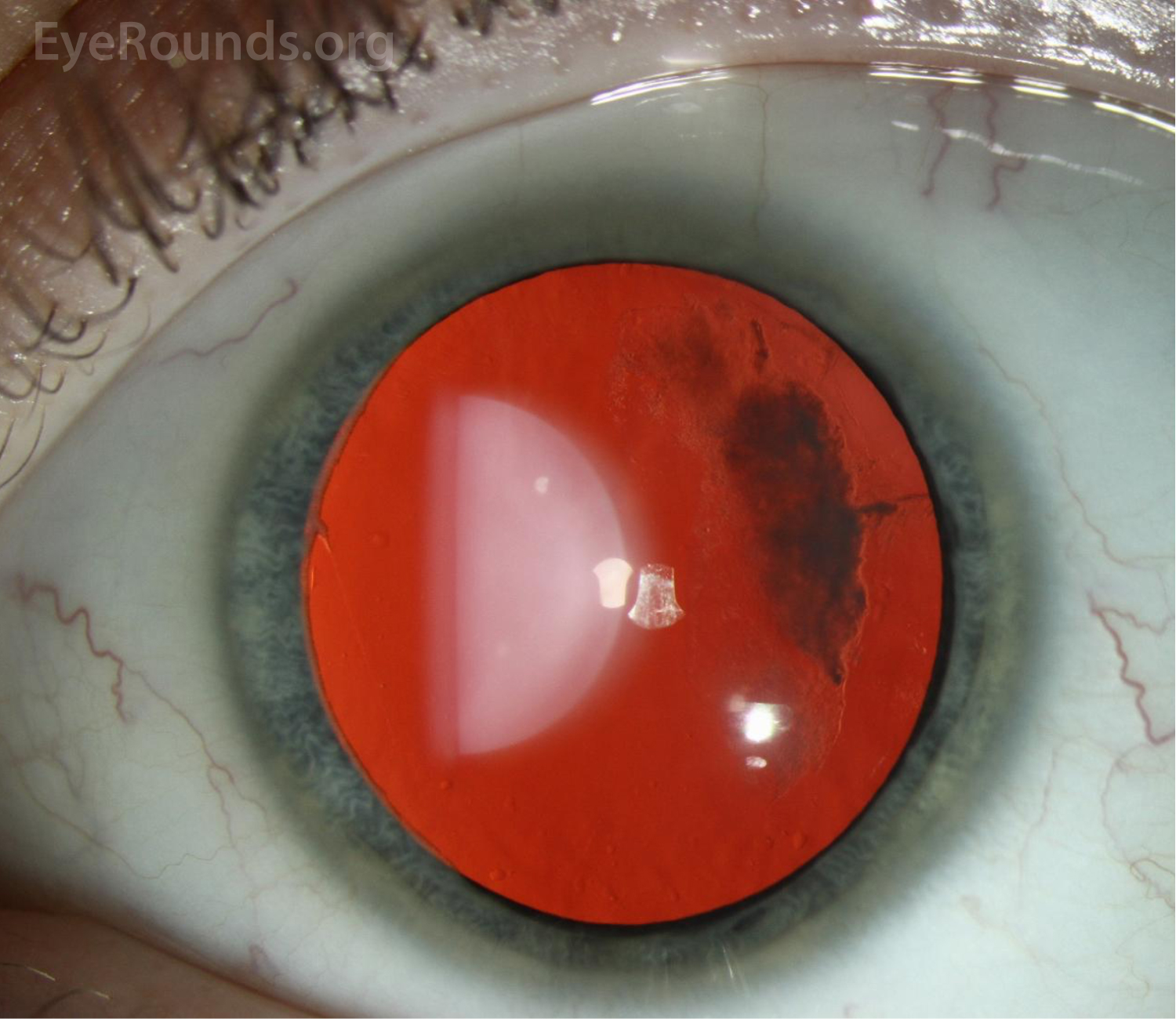
The patient was followed annually for routine dilated fundus exams. At age 13, he developed a macula-on retinal detachment in the right eye that was initially repaired by combined scleral buckle and pars plana vitrectomy with silicone oil tamponade. However, he developed a recurrent retinal detachment in the right eye due to proliferative vitreoretinopathy (Figure 1), ultimately requiring multiple additional repairs. He also developed mixed-mechanism glaucoma, requiring two Baerveldt setons in the right eye. Unfortunately, despite these interventions, his vision declined to hand motion in the right eye.
At the time of initial surgery in the right eye, the left eye was treated with indirect laser to areas of prominent lattice degeneration. At age 19, he developed a retinal detachment in the left eye that was repaired by scleral buckle and has remained attached.
DIAGNOSIS: Stickler Syndrome (Type I)
DISCUSSION
Etiology
Stickler syndrome is a multisystem collagen disorder that can manifest with orofacial, ocular, auditory, and skeletal features. It was originally described as hereditary progressive arthro-ophthalmopathy in 1965 by Gunnar Stickler.[1] Stickler syndrome is typically inherited in autosomal dominant fashion, with locus and allelic heterogeneity in genes coding for fibrillar collagens type II, IX, and XI. These collagens are collectively expressed in vitreous, cartilage, nucleus pulposus, and the inner ear.[2] Stickler syndrome is most commonly associated with pathologic variants in the genes COL2A1, COL11A1, and COL11A2, subdividing Stickler syndrome into type I, type II, and type III, respectively. There are a wide variety of presentations, with the COL11A2 variant considered a non-ocular variant.[3] Three other variants have been recognized and are due to mutations in the genes COL9A1, COL9A2, and COL9A3. These may demonstrate an autosomal recessive pattern but are extremely rare and have only been identified in a handful of families.[4] Although highly variable phenotypically, Stickler syndrome is thought to exhibit complete penetrance.[2]
Variants
Type I Stickler syndrome, caused by heterozygous variants in the COL2A1 gene, is the most common type of the disease, representing an estimated 80-90% of cases. Type I Stickler syndrome is the most common inherited cause of rhegmatogenous retinal detachment in childhood with high incidence of retinal detachment resulting from giant retinal tear.[5] It may present with both systemic and ocular findings, or ocular findings alone, depending on the mutation.[2]
Type II, due to variants in COL11A1, is the second most common form of Stickler syndrome, accounting for 10-20% of cases. In addition to eye abnormalities, Type II is the most likely to result in hearing loss. Marshall syndrome has multiple phenotypic commonalities as a result of COL11A1 mutations and is considered by some to be a subtype of type II Stickler syndrome.
Type III, due to COL11A2 variants, is sometimes referred to as non-ocular Stickler syndrome because the collagen gene product is not expressed in the eye, but is expressed in the joints, ears, and face, resulting in patients exhibiting most of the non-ocular findings recognized in other types. It is considered by some to be the same condition as otospondylomegaepiphyseal dysplasia (OSMED).
Types IV-VI are associated with variants in COL9A1, COL9A2, and COL9A3, respectively. Other candidate genes have been identified, including LOXL3 and LRP2,with presentations similar to a Stickler syndrome phenotype.[2]
Signs/Symptoms
Ocular manifestations have been recognized in Type I, Type II, and the more rare variants of Stickler syndrome. Findings frequently include vitreous structural abnormalities and may also include astigmatism, high myopia, retinal detachment, lattice degeneration, glaucoma, and cataracts, as seen in Figure 2.
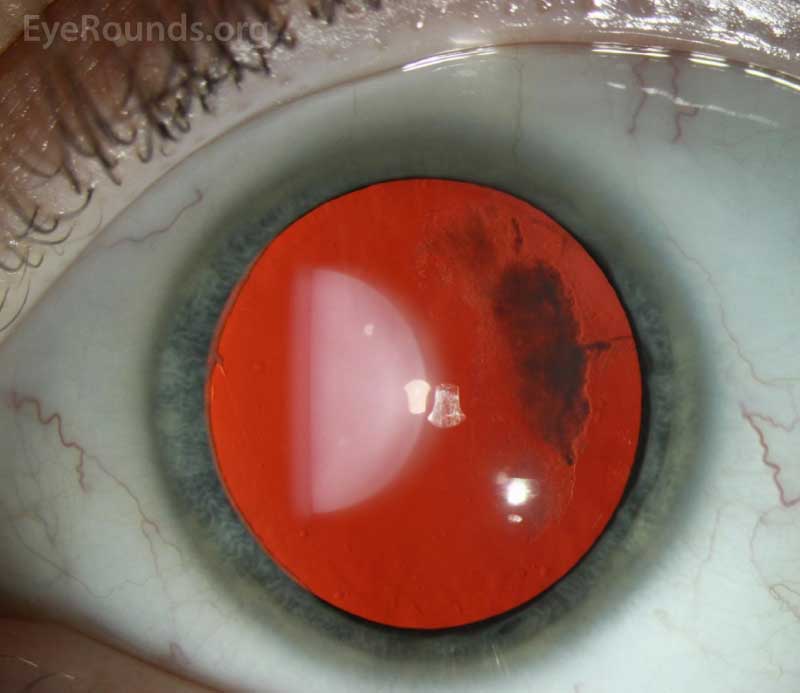
Vitreous abnormalities vary with the type of Stickler syndrome, sometimes described as membranous, beaded, or hypoplastic. Type I characteristically displays an optically empty-appearing central vitreous cavity with a membranous vitreous component within the vestigial gel in the retrolental space, as shown in Figure 3. Type II Stickler syndrome typically displays a beaded vitreous with irregular, thickened lamellae. Type II may less commonly have a hypoplastic vitreous which appears optically empty or with irregular lamellae architecture.[3]
Of note, patients with Wagner syndrome (as was initially suspected by the referring provider in this case) may also have optically empty-appearing vitreous. Patients with Wagner syndrome possess a pathologic variant in VCAN, which encodes versican, a protein involved in maintaining vitreous structure.[5] Although clinical features can be similar to Stickler syndrome, including myopia, early cataract formation, and retinal thinning, Stickler syndrome has a much higher prevalence of rhegmatogenous retinal detachment.
Frequent retinal detachments are particularly concerning for vision loss in Stickler patients and portend a poor visual prognosis. Retinal detachments occur most frequently between the ages of 10-30 years old and often require repeated surgical interventions due to high recurrence rates and proliferative vitreoretinopathy (as described in the case above).
Type 3 Stickler syndrome spares the eye. Individuals with Type VI without ocular symptoms have also been identified.
Other (non-ocular) features of Stickler Syndrome
Orofacial
One of the features common across the variants of Stickler syndrome is facial maldevelopment, namely a flattened facial appearance due to maxillary hypoplasia. Additionally, patients may have a depressed nasal bridge, anteverted nares, micrognathia, and midline clefting. These findings are usually most evident early in life and more subtle in adults. Pierre-Robin sequence is a particular constellation of craniofacial abnormalities that frequently co-occur in various diseases including Stickler syndrome that includes cleft palate, glossoptosis, and micrognathia.[6]
Auditory
Auditory abnormalities in Stickler syndrome may be sensorineural, conductive, or both. There is uncertainty in the pathogenesis for sensorineural hearing loss, but it is the most common auditory manifestation in Stickler syndrome. Conductive hearing loss has been observed and presumed due to either serous otitis media secondary to structural abnormalities of the syndrome including midline clefting and high arched palate, or structural ossicle defects.[6]
Skeletal
Skeletal abnormalities encompass a wide variety of presentations that are relatively nonspecific to Stickler syndrome. Many patients have spinal abnormalities, such as scoliosis, kyphosis, and platyspondyly, and complain of chronic back pain. Children may exhibit bony joint enlargement resulting from epiphyseal dysplasia. Many patients also have joint hypermobility with resulting complications and early osteoarthritis.[7]
Diagnosis
The diagnosis of Stickler syndrome is largely based on clinical features. Diagnostic criteria for Type I Stickler syndrome have been proposed (Table 1) based on observed clinical features, family history, and genetic findings.[8] In order to fulfill these criteria, patients must meet at least five points with at least one major 2-point criterion, and no more than 2 points may be assigned per major category (orofacial, ocular, auditory, skeletal).
| Abnormalities | |
|---|---|
| Orofacial |
|
| Auditory |
|
| Skeletal |
|
| Family History/Molecular Data |
|
While these criteria offer guidelines for diagnosis, they are not comprehensive for all forms of Stickler syndrome. They are limited in their application to patients with fewer involved systems, including patients with Type I who only have ocular manifestations. In an individual in whom Stickler syndrome is suspected, molecular testing should be performed to support the clinical diagnosis. If abnormalities are found in the recognized genes, it may help identify risks for other abnormalities that can be tested for, such as skeletal imaging, eye exam, and hearing tests. Results of molecular testing may also serve to inform patient of familial risks or help identify other genetic variants of Stickler syndrome.
Management
Treatment for Stickler syndrome consists of managing the manifestations of the disease as they develop. Surgical intervention may be required for oropharyngeal problems such as cleft palate. Ocular surgeries may include cataract removal and retinal detachment repair. Hearing aids, speech therapy, ear tubes, etc. may be useful in cases with involvement of the auditory system. Pain relief and physical therapy are often a critical aspect of Stickler syndrome management due to musculoskeletal problems including early osteoarthritis. Finally, lifestyle is an important consideration for joint stress and retinal detachment risk. Patients are often advised to be cautious in activities and to avoid contact sports.
In some cases, prophylactic interventions such as cryotherapy, argon laser photocoagulation, and scleral buckle can be considered due to the high lifetime risk of retinal detachment. However, prophylactic intervention remains controversial, as there are currently no clear guidelines on the best clinical context for their application, nor is there consensus on the benefits relative to their associated risks.[9]
Differential Diagnosis
ETIOLOGY
|
SIGNS & SYMPTOMS
|
VARIANTS
|
TREATMENT/MANAGEMENT
|
Sevcik K, Ramirez D, Han IC, Binkley EM, Han IC. Stickler Syndrome. EyeRounds.org. January 20, 2022. Available from https://EyeRounds.org/cases/320-stickler-syndrome.htm

Ophthalmic Atlas Images by EyeRounds.org, The University of Iowa are licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License.

Address
University of IowaLegal
Related Links

{kind=link}
{kind=link}
{kind=link}