Chief Complaint: Drooping of the right upper eyelid
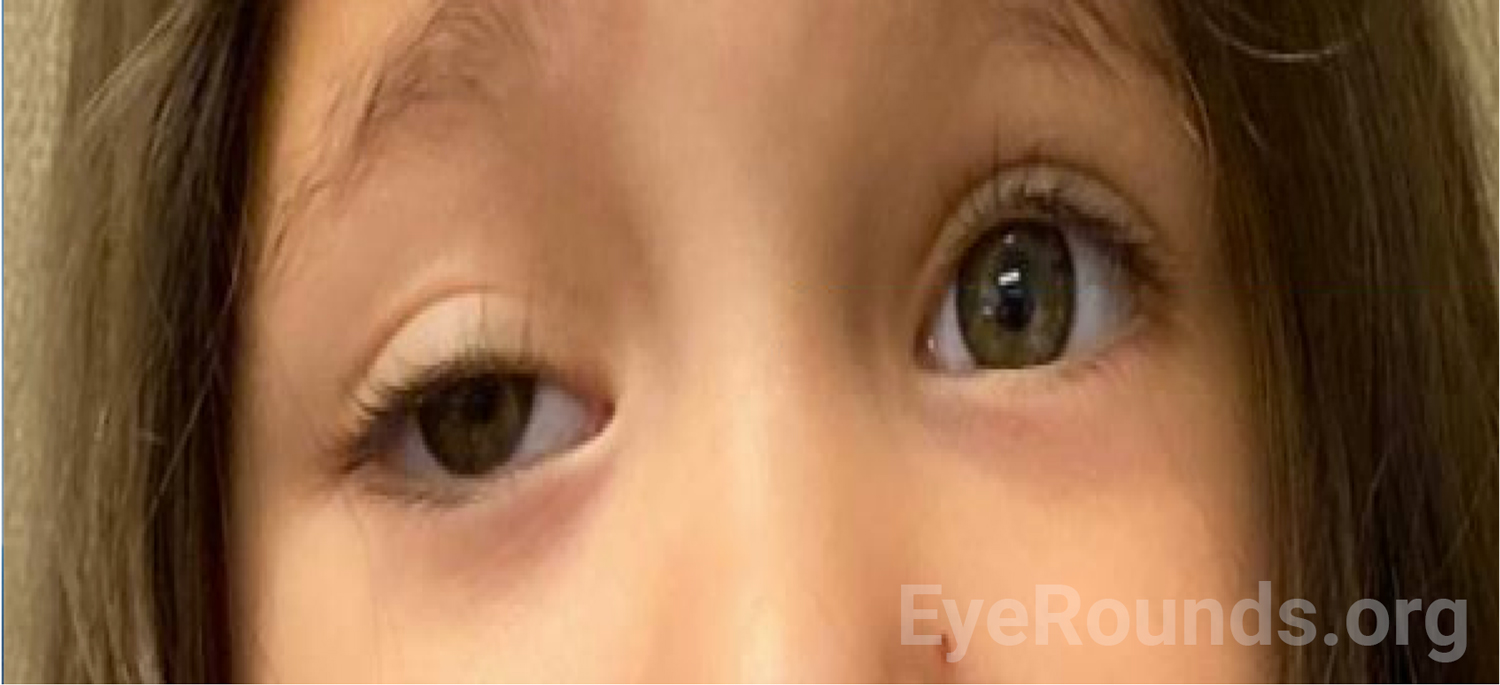
History of Present Illness (HPI): A 3-year-old Hispanic female with a history of retinopathy of prematurity presented to the pediatric ophthalmology clinic with a chief complaint of drooping of the right upper eyelid (Figure 1). The symptoms started 5 months prior, seemingly after she fell and broke her left clavicle. Around this time, her right eye also started to drift outwards. Her mother denied other concerns about the patient’s vision, and the patient did not display symptoms suggestive of pain or double vision.
She had no other muscle weakness, including no difficulty chewing or swallowing, change of voice, or facial droop. She was born premature at 27 weeks’ gestation.
Past Ocular History
Past Medical History
Medications
Allergies
Family History:
Social History:
Review of Systems:
OCULAR EXAMINATION
| OD | OS | |
|---|---|---|
| Lids/Lashes | Ptotic upper lid, margin reflex distance (MRD) 1 = -1, no obvious lid fullness, non-tender | Normal |
| Conunctiva/Sclera | Clear and quiet | Clear and quiet |
| Cornea | Clear | Clear |
| Anterior Chamber | Deep and quiet | Deep and quiet |
| Iris | Normal architecture | Normal architecture |
| Lens | Clear | Clear |
| Vitreous | Normal | Normal |
| OD | OS | |
|---|---|---|
| Disc | Pink, sharp border | Pink, sharp border |
| Macula | Normal | Normal |
| Vessels | Normal | Normal |
| Periphery | Normal | Normal |
Differential Diagnosis
CLINICAL COURSE
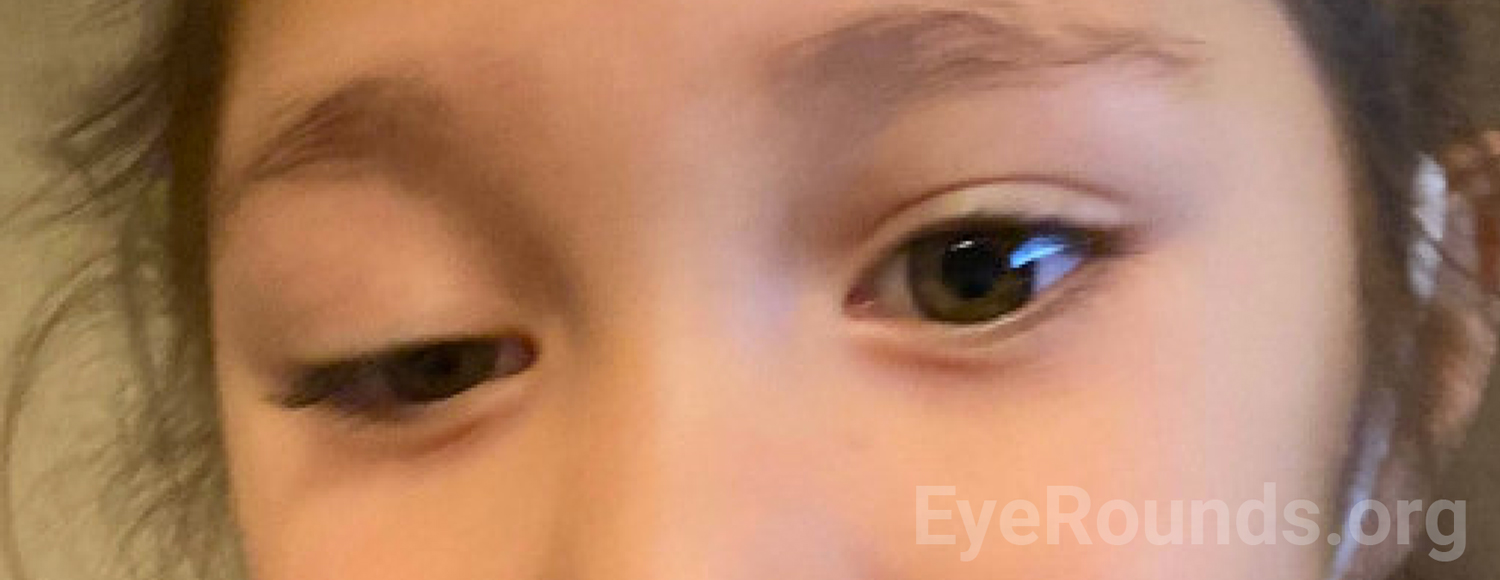
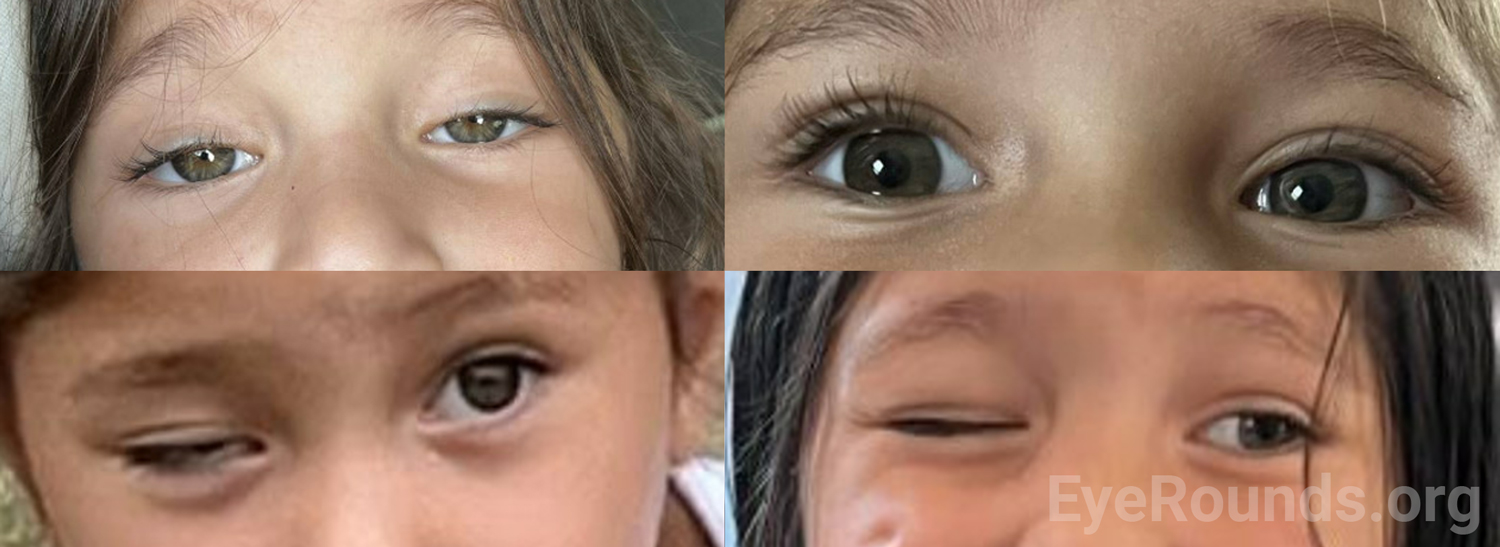
Given her history of trauma, the wide differential, and the goal of limiting anesthetic events, broad imaging was ordered to assess her sympathetic system, CN III course, and orbit. MRI of brain and MRA of neck with and without contrast showed no intracranial mass lesion, no cervical internal carotid artery dissection, no posterior communicating artery aneurysm, and no mass lesion along the bilateral cervical internal carotid arteries or skull base. MRI of orbit with and without contrast showed normal extraocular muscles and normal optic nerves bilaterally. At her follow-up visit in the eye clinic two months later, she had fluctuating ptosis of both upper eyelids (right>left) more significant towards the end of the day and assumed a chin up position when she was tired (Figure 2 and 3). A sensory motor examination showed intermittent right exotropia. Given her confirmed fluctuating symptoms of ptosis now affecting both upper eyelids and intermittent exotropia, suspicion was high for myasthenia gravis. Acetylcholine receptor (AChR) antibody level came back elevated and she was referred to a pediatric neuromuscular specialist.
Physical exam performed by the pediatric neuromuscular specialist showed right sided ptosis with eyelid covering her pupil to mid position. No Cogan lid twitch was noted with prolonged downward gaze. She was not cooperative for prolonged upgaze or ice pack testing. Her neuromuscular exam was otherwise normal.
Given her clinical presentation and laboratory work-up, she was diagnosed with juvenile ocular myasthenia gravis and was started on 12 mg (.87mg/kg) of pyridostigmine every 6 hours. She had improvement in ptosis initially after taking pyridostigmine but relapsed after a few weeks. At a follow-up visit two months later, her ocular exam showed persistent ptosis of the right eye and intermittent exotropia stable from the last visit. The dose and frequency of pyridostigmine was increased to 18mg (1.15mg/kg) every 4 hours to better manage her symptoms.
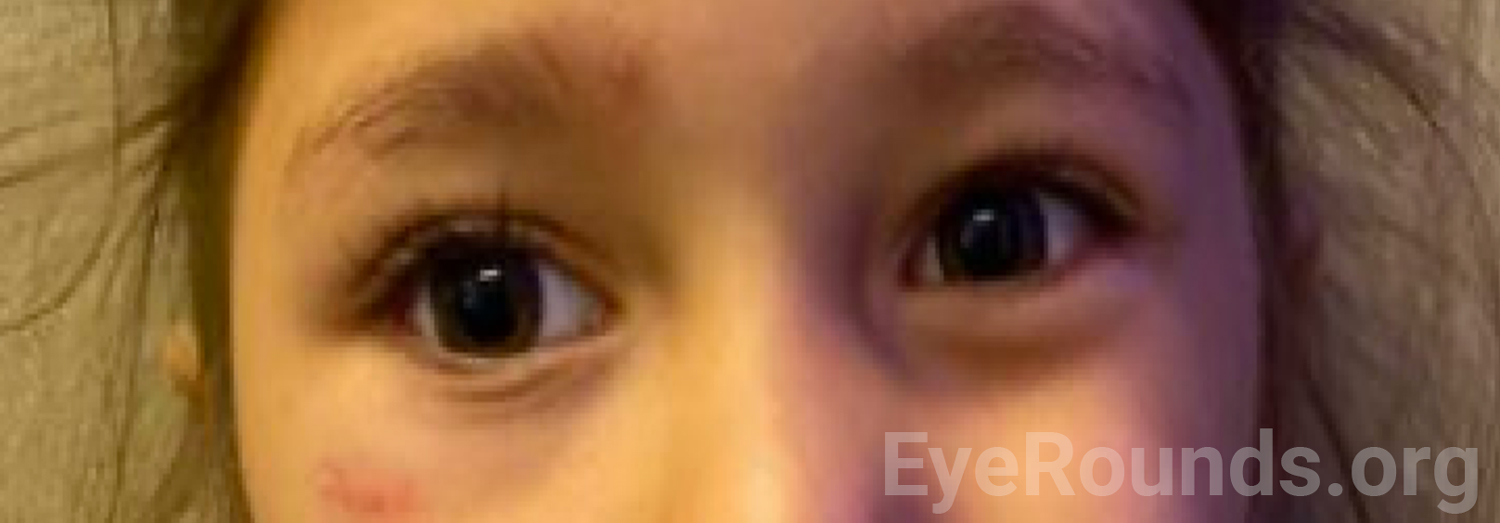
At her most recent follow-up visit three months later, she had significant improvement in symptoms with only mild ptosis of her right upper eyelid (Figure 4). She was continued on 18 mg every 4 hours of pyridostigmine. Imaging of her thymus is planned to rule out thymoma. She continues to follow in the eye clinic to monitor for any signs of amblyopia and to manage her intermittent exotropia, while pediatric neurology is managing her medication.
DIAGNOSIS: Juvenile ocular myasthenia gravis
DISCUSSION
Etiology/Epidemiology
Juvenile myasthenia gravis (JMG) is an autoimmune neuromuscular disorder resulting in weakness and fatigability of skeletal muscle in infants, children, and adolescents aged 0-18 years [1]. Ocular JMG is localized to the levator palpebrae superioris, orbicularis oculi, and extraocular muscles [1] and is defined as having isolated ocular symptoms for two years without generalization of symptoms [3]. Since patients usually present with isolated ocular symptoms, differentiation between generalized versus ocular myasthenia gravis is not possible at symptom onset [2,3].
Ocular JMG accounts for 10-35% of cases of MG in childhood [4,5] with a higher proportion of children with ocular JMG reported in Asian populations [6,7]. Although spontaneous remission in prepubertal patients is frequent, it has been estimated that 50% of children with ocular symptoms develop generalized myasthenia within 2 years of symptom onset [1]. Risk factors for generalization of MG in the adult population have been noted to include acetylcholine receptor antibody positivity (also noted in the pediatric population [8], presence of thymoma, a positive decrement on repetitive nerve stimulation study, and presence of other autoimmune disorders [2-5].
Pathophysiology
Myasthenia gravis (MG) is a B-cell mediated autoimmune disorder of neuromuscular transmission. The majority of MG is characterized by antibodies directed towards the acetylcholine receptors (AChR) at the post-synaptic membrane of the neuromuscular junction [6,7]. These pathogenic antibodies induce loss of the AChR through several mechanisms, including: complement mediated destruction of the motor end plate [8], internalization and degradation of the AChR, and direct interference with ACh binding to the receptor [9,10]. A less frequent form of MG includes MuSK antibodies, but the pathogenic mechanism of this form is less clear [6,10-12].
It is unclear why eye muscles are so frequently involved in MG, but several mechanisms have been proposed [13,14]. The levator palpebrae superioris is under constant activation during eye opening and may be particularly susceptible to fatigue. Furthermore, subtle alterations in fine movements of extraocular muscles are more likely to produce noticeable symptoms compared to limb muscles [13,14]. In addition, the unique immunological environment of orbital tissues may increase their susceptibility to autoimmune disorders such as MG [15].
Signs/Symptoms
Ocular symptoms are the most frequent clinical manifestation of JMG with a recent meta-analysis showing that ptosis, diplopia from strabismus were the first clinical manifestations of JMG in 60.6% of patients [16]. The most common ocular symptom was ptosis, which occurred in 77.7% of JMG patients who initially presented with ocular symptoms [16]. Generalized muscle weakness at initial presentation is rare in JMG [7,16].
Ocular symptoms are characterized by fluctuating and fatigable weakness aggravated by increased physical activity or repetitive use. Symptoms may improve with rest, sleep, and cold temperature. Ocular manifestations may result in a constricted superior visual field from upper eyelid ptosis and diplopia from strabismus [6]. Children may complain of double vision towards the end of the day, have difficulty climbing steps due to impaired depth perception, or adopt a chin-up head position due to ptosis [1].
Testing/Laboratory work-up
The diagnosis of JMG is primarily clinical, but positive serology and neurophysiologic testing can support the diagnosis [17].
Physical exam findings
Eyelid fatigue can be elicited on physical exam through sustained upgaze for 1-2 minutes. Worsened ptosis upon return to primary gaze is a positive finding. The Cogan lid twitch sign is also a positive finding, which is elicited after instructing the patient to shift from downgaze to primary gaze with an affected eyelid quickly rising and falling by as little as 1mm [18]. Improvement of ptosis after resting the eyelids for 5-8 minutes by closing the eyes is also a supporting sign.
Ocular myasthenia gravis (OMG) can impact one or multiple extraocular muscles in either or both eyes, and diplopia caused by ocular misalignment from ophthalmoparesis should be assessed by testing the cardinal positions of gaze. Fatigable ophthalmoparesis after prolonged or sustained gaze in the direction of action of an involved muscle is a positive sign [19]. In addition to ptosis and ophthalmoparesis, other findings may include lagophthalmos due to orbicularis oculi weakness.
The ice pack test is based on the principle that neuromuscular transmission is improved at a lower temperature and has a sensitivity of 80% and specificity of 100% in those with prominent ptosis [20,21]. The patient is instructed to place a bag of ice on the eyelid for one minute, and extent of ptosis is assessed after removal [20,21].
Serum antibody studies
Positive serology of autoantibodies targeting AChR in JMG ranges from 70-80% in cohort studies and is less commonly positive compared to adult MG [17]. Positive serology for AChR antibodies in ocular JMG compared to generalized JMG is even less common [22,23], but it should be noted that delayed seroconversion can occur up to 5 years after onset, so seronegative JMG patients should be tested regularly at 6 month intervals [17]. Commercially available tests, including the radioimmunoprecipitation (RAI) assay and the enzyme linked immunosorbent assay (ELISA), can be used to check serology [17]. MuSK antibodies account for 5-8% of all MG patients and can be tested for if patients are seronegative for AChR antibodies [24].
Neurophysiology
Neurophysiologic studies can be an important supplement to immunologic studies [17]. Repetitive nerve stimulation (RNS) and single-fiber electromyography are known diagnostic tests for myasthenia gravis, but they can be technically challenging in young children [17] and are not as sensitive in patients with OMG [25,26]. Single-fiber electromyography [27] is more sensitive than RNS in screening for OMG [28].
Imaging
All children with JMG should have chest imaging (CT or MRI) to assess thymoma associated with their MG [13]. Imaging studies of the brain and orbit can exclude treatable inflammatory structural disease or brainstem or cranial nerve abnormalities in the initial work up for ptosis and diplopia [25-27].
Treatment/Management
There are no standardized or internationally accepted guidelines for treatment of JMG or ocular JMG, but management should include a multidisciplinary approach with specialists from pediatric neurology and ophthalmology [13]. Goals of treatment are two pronged: symptom management and immunomodulatory treatment.
Cholinesterase Inhibitors
Cholinesterase inhibitors (ChE-I) are the first line treatment for symptom management of JMG and work by inhibiting the breakdown of acetylcholine at the neuromuscular junction [13]. Pyridostigmine is a non-selective ChE-I and is the most widely used ChE-I for JMG [13]. The dose of pyridostigmine should be titrated according to symptom relief and side effects experienced. Although slow-release forms of pyridostigmine are available, their high dose preparations limit use in children [13]. If symptoms persist with doses of 1mg/kg 4 times per day, other options should be explored such as immunosuppressive agents [13].
No clinical trials have been conducted evaluating the impact of ChE-I in OMG [28], but case series show pyridostigmine treatment alone rarely results in complete resolution of ocular symptoms, especially diplopia, with only 6.9% of OMG patients showing resolution of primary gaze diplopia [29].
Immunosuppressive agents
There are no formal guidelines for the use of immunosuppressive agents in JMG, but corticosteroids are widely accepted as the first line agent [30]. The goal of immunosuppressive therapy is to induce remission and slowly wean off of ChE-I and then steroids [13]. In patients who do not have spontaneous remission, steroids are often required for complete resolution of diplopia and ophthalmoparesis [31], but the potential side effects should be carefully weighed against the benefits as approximately 10% of patients will have spontaneous remission without them, especially in pre-pubertal patients [32].
Steroid-sparing agents such as azathioprine, mycophenolate mofetil, cyclosporine, rituximab, and intravenous immunoglobulin are second-line immunotherapeutic agents for patients who require high-dose steroid treatment or cannot tolerate its side effects [33].
Thymectomy
High rates of thymic pathology are seen in MG patients, and all children with JMG should have thymic imaging (CT or MRI) regardless of clinical presentation [13]. In adults, if a thymoma is present, thymectomy is indicated [13]. The role of thymectomy in non-thymomatous MG is dependent on several factors, and its role in JMG is unclear as no clinical trials have been conducted in pediatric patients [13]. A recent cohort study showed that among 488 patients with JMG (half of which had pure ocular symptoms) who underwent thymectomy, 77% of patients symptomatically improved, and 29% experienced sustained remission [34]. The role of thymectomy in children with milder disease is less clear given the high rates of spontaneous remission, especially in pre-pubertal children [13], but it should be considered in AChR-Ab positive ocular JMG patients to minimize long-term complications of immunosuppressive treatments, such as steroids [13].
Additional management of ophthalmic symptoms
Children with ocular JMG require close monitoring by an ophthalmologist due to the risk of amblyopia from ptosis or strabismus [2]. Diplopia can be managed with an eye patch, Bangerter occlusion foil, or prism. It is estimated that 25-50% of patients with JMG may develop amblyopia [35,36]. If treated aggressively, rates of residual amblyopia in patients with JMG can be reduced to 3% [36]. As such, patients should be monitored carefully for amblyopia resulting from strabismus and ptosis and treated accordingly. Ptosis may be present following stabilization of disease in 46-66% of patients [35,36]. Strabismus surgery and ptosis surgery can be considered once disease has stabilized [37].
EPIDEMIOLOGY OR ETIOLOGY
|
SIGNS
|
SYMPTOMS
|
TREATMENT/MANAGEMENT
|
RELATED CASES
Ajjarapu A, Strampe MR, Thurtell MJ, De Andrade LMTorpedo Maculopathy. EyeRounds.org. Posted January 4, 2024; Available from https://EyeRounds.org/cases/353-juvenile-ocular-myasthenia-gravis.htm

Ophthalmic Atlas Images by EyeRounds.org, The University of Iowa are licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License.

Address
University of IowaLegal
Related Links