INITIAL PRESENTATION
Chief Complaint: Intermittent blurry vision in a patient with history of walking issues, spasticity, and hemiplegia
History of Present Illness
A 66-year-old patient with a history of left eye childhood amblyopia, was referred to the neuro-ophthalmology clinic by the genetic service after she was found to have a mutation reportedly related to optic atrophy.
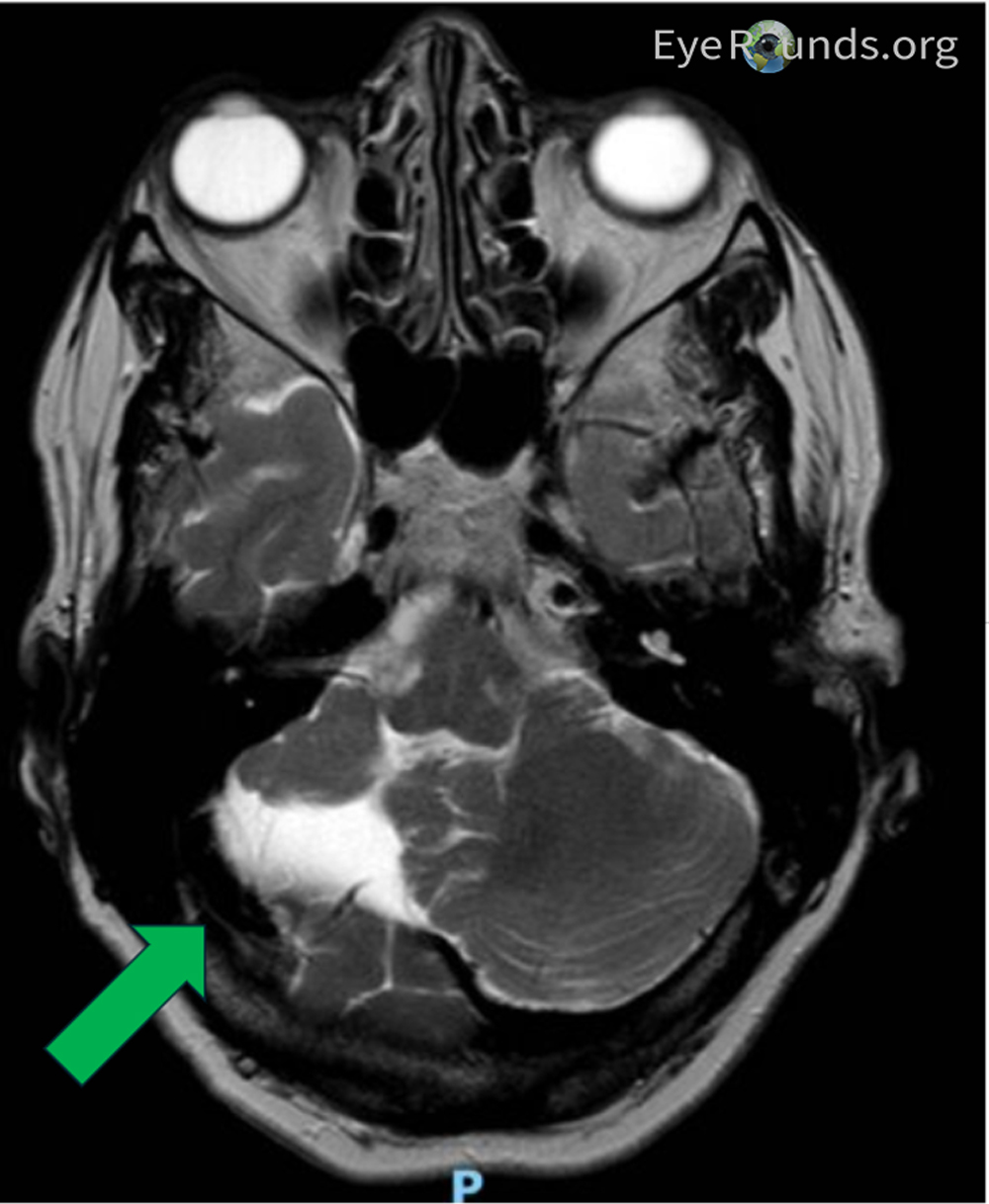
She started developing issues walking and had spasticity starting at age 49 and was eventually diagnosed with cerebellar hypoplasia at age 50 based on brain MRI (Figure 1). She progressively lost her ability to walk due to lower extremity spasticity and began to use a wheelchair and a walker. Her mother and maternal grandmother also had adult-onset difficulty walking requiring assistance. At age 63, she had cataract surgery in both eyes with good outcome, but 1 year afterwards, she began to have intermittent blurry vision in both eyes, that did not improve with YAG laser capsulotomy. No abnormalities of her fundus or nerves were noted at that time. Eventually, a consultation with the neuro-ophthalmology service was requested.
Past Ocular History
Past Medical History
Past Surgical History
Medications
Allergies
Family History
Social History
Review of Systems
OCULAR EXAMINATION
| OD | OS | |
|---|---|---|
| Lids/lashes | Normal | Normal |
| Conjunctiva/sclera | White, quiet | White, quiet |
| Cornea | Clear | Clear |
| Anterior chamber | Deep and quiet | Deep and quiet |
| Iris | Normal architecture | Normal architecture |
| Lens | Intraocular lens implant | Intraocular lens implant |
| Anterior vitreous | Normal | Normal |
| OD | OS | |
|---|---|---|
| Vitreous | Posterior vitreous detachment | Posterior vitreous detachment |
| Disc | Marked diffuse pallor | Marked diffuse pallor |
| Cup-to-disc (C/D) ratio | 0.8 | 0.8 |
DIAGNOSIS: Spastic Paraplegia Type 7 (SPG79A) associated Optic Neuropathy
CLINICAL COURSE
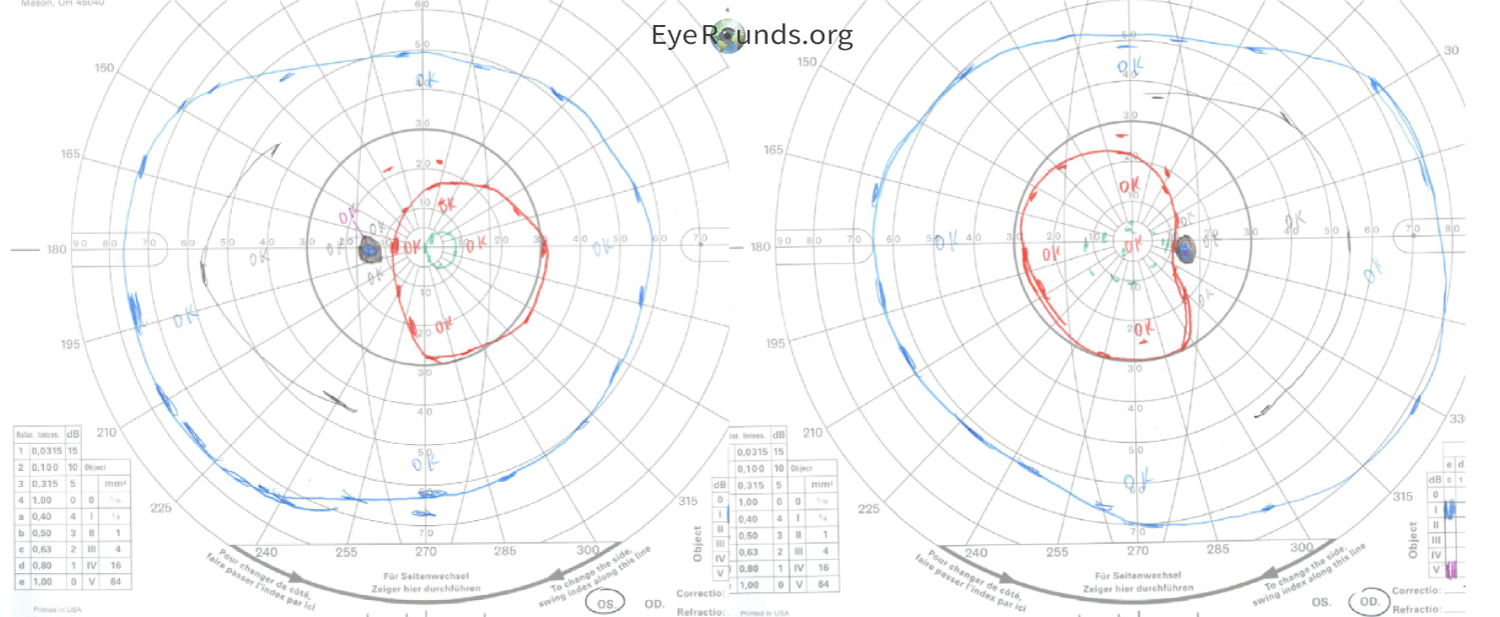
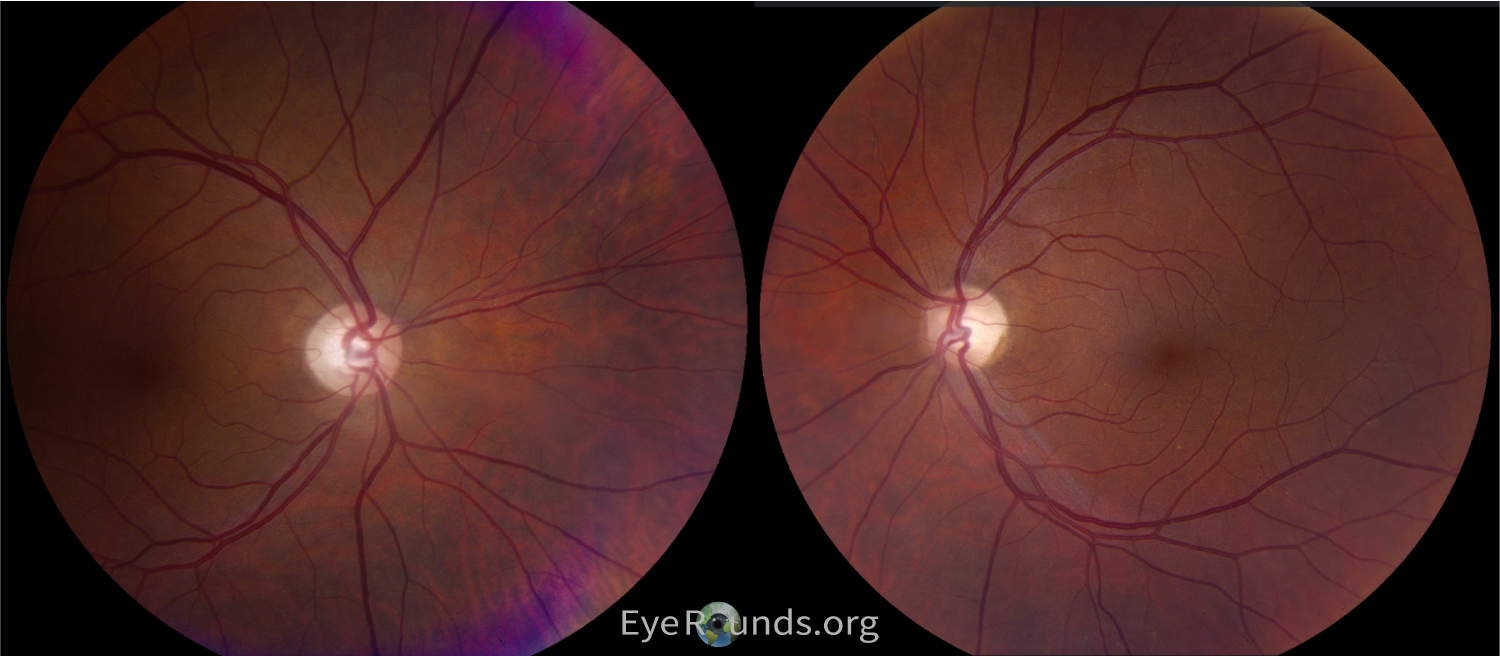
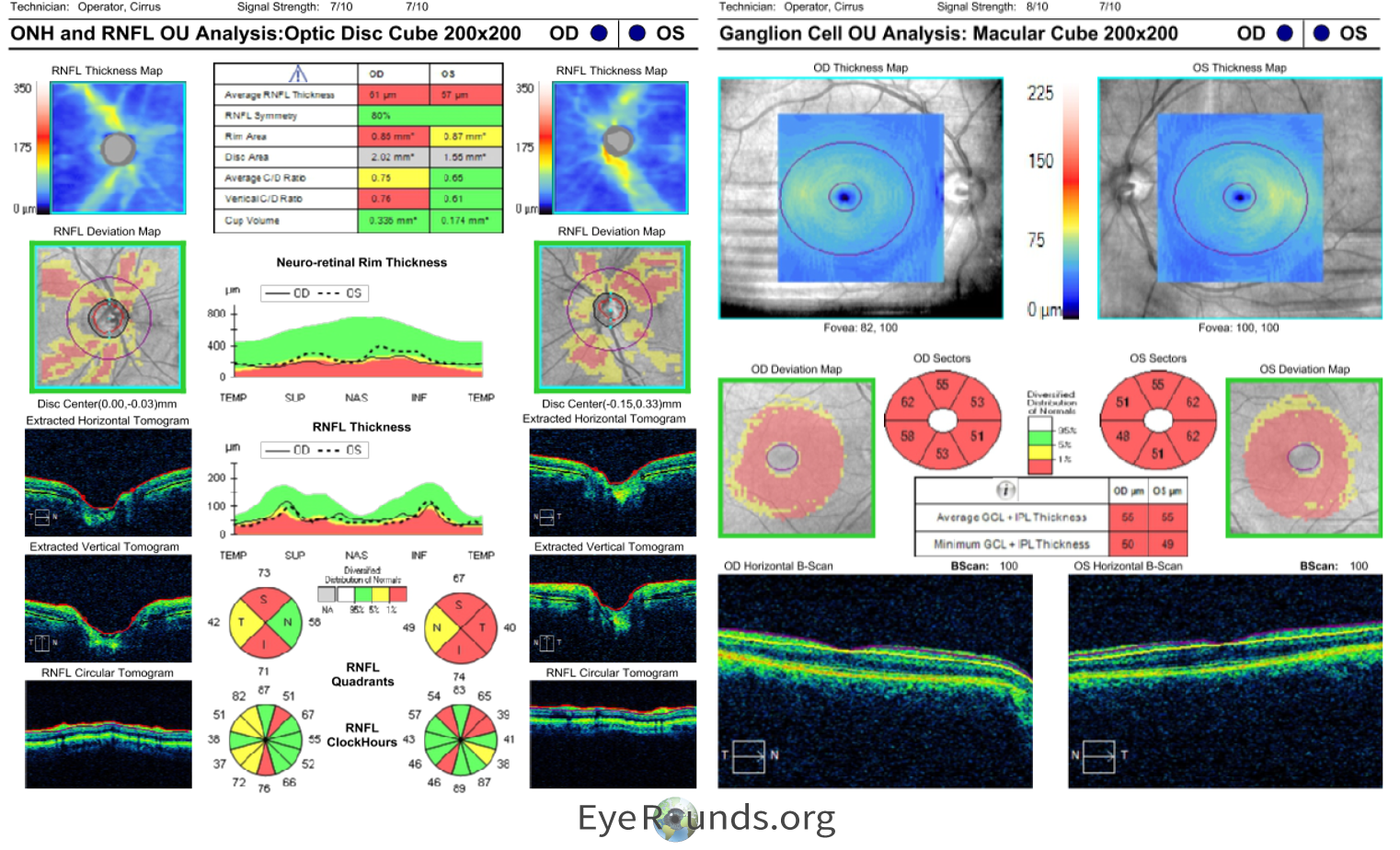
The patient underwent genetic testing which revealed a single heterozygous pathogenic variant within the UCHL1 gene: c.532C>T (p.Arg178*). She was given a genetic diagnosis of SPG79A, denoting the autosomal dominant version of this condition. She was given a clinical diagnosis of hereditary spastic paraplegia (HSP), which was noted to be related to optic atrophy. This appeared to be autosomal dominant based on family history, as her mother, maternal grandmother, and one of her brothers had adult-onset problems with walking without a causative diagnosis. Nobody else in her family had opted to take the genetic testing. She did not know about her diagnosis at the time of her cataract surgery, and the cause for her intermittent blurry vision was not found until her genetic testing and neuro-ophthalmic workup and testing. Her left eye amblyopia was the likely cause of her 20/40 vision, but she was visually functioning well overall. Her testing showed depression of the central isopters of both eyes on Goldmann Visual Field (Figure 2), diffuse optic disc pallor of both eyes (Figure 3), and RNFL and GCL thinning on OCT indicating a longstanding atrophic optic neuropathy (Figure 4). It is possible a pre-surgical OCT may have revealed optic atrophy in the patient at an earlier timepoint.
The patient was educated about the findings. She was already on coenzyme Q10 supplements, and was recommended to avoid smoking and alcohol consumption, prioritize adequate dietary/nutritional intake, and to avoid unnecessary metabolic and oxidative stress. She was to follow up with Neurology and Genetics as planned and to return to neuro-ophthalmology once yearly for monitoring.
DISCUSSION
Etiology/Epidemiology
Hereditary spastic paraplegia (HSP) is a heterogeneous group of genetic neurodegenerative diseases first described in 1880 (1,2). With over 80 genes implicated thus far, clinical manifestations are wide-ranging. The prevalence varies between 0.1 and 9.6 per 100,000 depending on the subtype (3). Early-onset HSP is defined as onset of 35 years or under, while classic and late onset are above 35 years of age (3).
A subtype of HSPs includes spastic paraplegia 79A (SPG79A), which is an autosomal dominant form of HSP with ataxia. The onset is usually in adulthood with the median being 49 years. SPG79B is a recessive form that presents similarly except with an earlier onset and slightly more severe symptoms. Historically, SPG79 has been linked to recessive mutations, but recent data have delineated the two with SPG79A being the dominant form (4). Both are mutations at chromosome 4p13 in the ubiquitin C-terminal hydrolase L1 (UCHL1) gene. The exact functions of UCHL1 are not fully understood, though it is thought to be a ubiquitin-processing enzyme necessary for axonal health and stability (5).
Spinocerebellar ataxia (SCA) is a group of similar progressive neurodegenerative diseases with a global prevalence of 1-5 per 100,000. Unlike HSPs, mutations in SCA are more often related to CAG nucleotide repeats that result in toxic aggregation of proteins (6).
HSPs can be differentiated from other hereditary optic neuropathies with progressive vision loss such as dominant optic atrophy (DOA) and Leber’s hereditary optic neuropathy (LHON). Both are rare conditions, but they have earlier peak onsets in the first decade or second and third decades, respectively (7,8). DOA has a global prevalence of 1 in 30,000 while LOHN has a global prevalence of 1 in 50,000 with 4-5x more cases in males compared to females. DOA and LHON are linked to mutations in the OPA and MT-ND genes, respectively, which all encode mitochondrial membrane proteins.
Pathophysiology
HSP is a very heterogenous disease with numerous causative mutations. The clinicopathologic manifestations slowly progress with higher challenges to diagnose early. The HSP subtype SPG79A observed in this patient results from the substitution of arginine 178 for a premature stop codon in the Uchl1 protein, one of the most abundant in the human brain (10). The UCHL1 protein stabilizes ubiquitin monomers, increasing their availability to be tagged onto proteins destined for proteasomal degradation (11). Consequently, it is hypothesized that mutated UCHL1 causes neuronal dysregulation by two main mechanisms: via impaired ability to reliably mark proteins for degradation by the proteosome, and subsequent toxic aggregation of protein products (12).
UCHL1 dysregulation has been associated with other multisystem neurodegenerative syndromes including Parkinson disease, cerebellar atrophy, dorsal column dysfunction, nystagmus, blindness, and upper motor neuron dysfunction (13). MR imaging in patients with UCHL1 mutations display optic chiasm, optic nerve, and optic tract atrophy, and diffuse tensor imaging shows Wallerian degeneration of the optic radiations (13).
Signs/Symptoms
HSPs are primarily characterized by axonal atrophy in the spinal cord leading to lower extremity weakness and paresthesia, hypertonicity, and bladder disturbances. (2) Specifically, SPG79A presents with ataxia, lower extremity spasticity, and sensory neuropathy. (4) Ocular manifestations are common which include vision loss, optic atrophy, and saccadic eye movements.
Optic atrophy is not an uncommon manifestation of HSP and can occur in several subtypes. For example, the most common autosomal recessive HSP, SPG11, accounts for 8% of all HSP cases with reported cases of visual failure secondary to optic atrophy (2). SPG7 is another autosomal recessive subtype where ocular signs and symptoms can occur including progressive external ophthalmoplegia, visual loss secondary to optic neuropathy, and subclinical abnormalities in OCTs. Of note is that both subtypes can present with manifestations on neuroimaging; for example, an estimated 39-95% of SPG7 cases have cerebral atrophy on MRI (2).
Symptoms of SCA tend to present later in life with signs and symptoms related to abnormalities of cerebellum, but other central and peripheral nervous system signs are common as well. For example, SCA1, SCA2, and SCA3 all commonly present with problems with ocular motility, nystagmus, and exophthalmos. Additionally, SCA7 can present with initial cone and later rod dystrophy leading to vision loss (6).
HSPs can be differentiated from DOA and LHON by clinical course as these optic neuropathies present earlier in life (7). People with non-syndromic DOA can have later presentations which include signs and symptoms such as bilateral, symmetric disc pallor and central scotomas. Syndromic DOA has more severe visual deficits, and in later stages of life, manifestations such as motor and sensory neuropathy, spastic paraplegia, and cerebellar ataxia. Unlike HSPs, vision loss is typically the only symptom in LHON, although other extraocular abnormalities have been reported (8).
Testing/Laboratory work-up
Genetic testing for HSP leads to greater accuracy and peace of mind for patients. Labs performing testing for HSP need to be CLIA-approved and provide estimates of variant pathogenicity (14). The preferred specimen for testing includes 3mL whole blood, and alternate specimens include saliva, buccal swab, and gDNA (15). Because HSP is largely untreatable, testing in asymptomatic minors is generally discouraged. The testing process itself involves collaboration between ophthalmologists, genetic counselors, and geneticists. Genetic counselors play a key role in understanding family history, selecting appropriate tests, and explaining results. While interpreting results can take time, patients should receive a copy of their report (2,14). Neuroimaging with MRIs and neurophysiologic testing including somatosensory evoked potentials, electromyography, and nerve conduction studies may be useful as abnormalities are common across many HSP subtypes (2).
Next-generation sequencing (NGS)-based gene panels for hereditary spastic paraplegia (HSP) have gained traction due to their increasing cost-effectiveness and widespread availability (2). These panels primarily target the exonic regions of a comprehensive set of HSP-associated genes. However, NGS gene panels often fail to detect copy number variations (CNVs) such as large deletions or duplications, including exon deletions. Additionally, mutations occurring in promoter regions, deep intronic sequences, and those involving triplet repeat expansions typically fall outside the scope of these panels (2). NGS panels for HSP typically include diseases which might present with a similar clinical phenotype as myelination abnormalities, neurometabolic defects, and spinocerebellar ataxias (2).
Treatment/Management
Currently, there are no specific, evidence-based treatment options for HSPs. Treatments are mainly symptom management with medications such as muscle relaxants and physical therapy to improve gait. So far, little attention has been paid to improving symptoms outside of spasticity and gait. A multidisciplinary spasticity clinic setting presents the optimal approach for comprehensive symptom management. This integrated environment allows for the targeted treatment of stiffness, cramps, spasms, and deformities associated with spasticity (2).
For SPG7, there is anecdotal evidence and case reports of usage of coenzyme Q10 supplementation, due to the potential reduction of reactive oxygen species-induced stress of the optic nerve. Avoidance of toxic triggers like smoking, alcohol, and illicit drugs may be helpful to recommend to patients to further preserve optic nerve tissue functioning, in addition to preventative measures with a healthy and balanced diet. Patients should have established care with neurologists, and genetic counseling should be offered to their families (2).
EPIDEMIOLOGY OR ETIOLOGY
|
DIAGNOSIS
|
SYMPTOMS/SIGNS
|
TREATMENT/MANAGEMENT
|
Chun LY, Young B, Zhu M, Rodriguez Leon A. Spastic Parapalegia. EyeRounds.org. June 26, 2024. Available from https://EyeRounds.org/cases/359-Spastic-Parapalegia.htm

Ophthalmic Atlas Images by EyeRounds.org, The University of Iowa are licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License.

Address
University of IowaLegal
Related Links