
Chief Complaint: 31-year-old female patient with gradual decrease in her visual field in the left eye (OS).
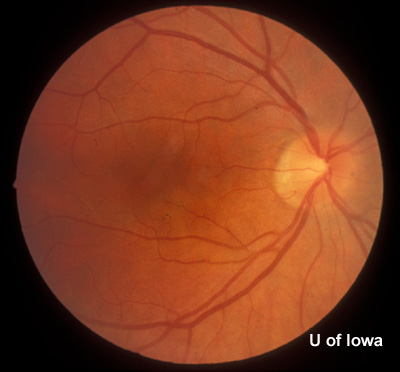
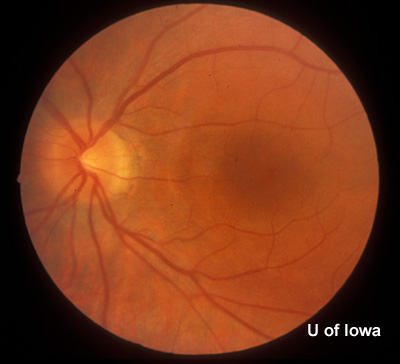
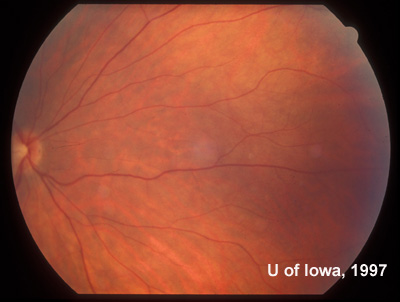
History of Present Illness: This patient was first seen at the University of Iowa Hospitals and Clinics (UIHC) Department of Ophthalmology at age 18 for simple myopia. Refraction at that time (1971) helped the patient attain a visual acuity of 20/15 in each eye. Fundus examination revealed the incidental finding of an area of congenital hypertrophy of the retinal pigment epithelium (CHRPE) in the periphery of the left eye. Fundus photography was obtained of the posterior and peripheral fundus shortly after this visit (See Figures 1 and 2).
| Figure 1A: Fundus, right eye (OD) | Figure 1B: Fundus, left eye (OS) |
 |
 |
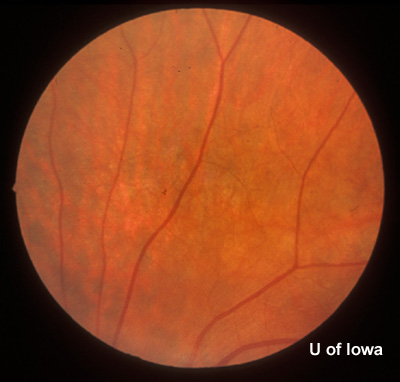
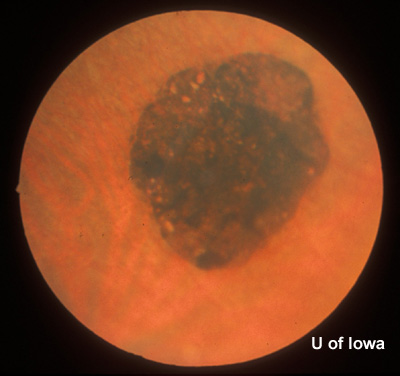
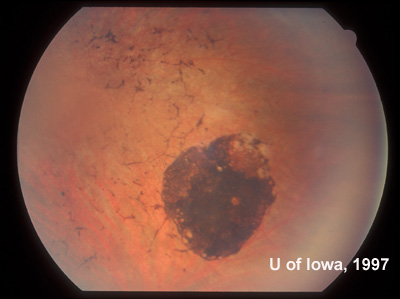
| Figure 2A: Peripheral fundus, OS | Figure 2B: CHRPE in peripheral fundus, OS |
 |
 |
The patient returned to UIHC for routine eye examinations over the next several years, all of which were normal. Fundus examinations in the 1970's revealed no other abnormalites in either the macula or peripheral fundus of either eye. From age 22 to 32 the patient received routine examinations and refraction from her local optometrist which were reportedly normal. During this ten year period, however, the patient began to notice gradual decrease in the peripheral field of the left eye. For this new complaint, the patient was referred back to UIHC in 1985. The findings from that visit are reported below:
Ocular History: Simple myopia, both eyes (OU). Small area of congenital hypertrophy of the retinal pigment epithelium (CHRPE) in the left eye, as noted. No previous eye surgery, inflammation, or any history of eye trauma.
Medical History: Unremarkable. Complete review of systems did not reveal any history of inflammatory disease, prior infection, infantile illness or sexually transmitted disease.
Medications: None
Family and Social History: No family history of acquired or inherited eye disease. No history of diabetes. An extensive review of the family pedigree revealed no other family member in four generations with any vision problems other than ammetropia that was correctable with refraction.
Ocular Exam
 |
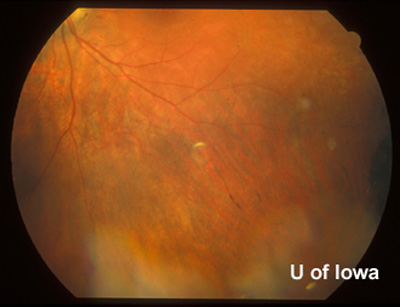
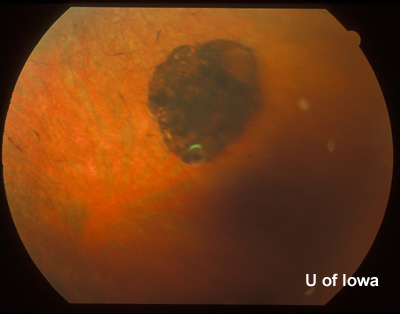
| Figure 4A: Peripheral fundus, OS. Small linear clumps of pigment and granular pigment changes. | Figure 4B: Peripheral fundus, OS. CHRPE, as before, and linear pigment changes. |
 |
 |
Course: At this point we considered the differential diagnosis for unilateral retinal pigment epithelium changes and peripheral field loss with preserved central vision, including:
Additional history was obtained and testing initiated to refine this differential. The patient had no history or evidence of trauma nor did she have a metallic intraocular foreign body. Examination and ultrasound over the CHRPE lesion further confirmed the absence of any occult material at the location of this classic congenital lesion. There was no evidence of prior retinal detachment, nor the segmental pigmentation changes that would be expected from detached retina. The patient had never had any serious systemic infection and all immunizations were up to date. Laboratory analysis for syphilis revealed a non-reactive rapid plasma reagin (RPR). The venereal disease research laboratory (VDRL) screening test and the fluorescent treponemal antibody absorption (FTA) test were also negative. Toxoplasmosis titers showed no evidence of prior infection and the patient's complete blood count (CBC) and electrolyte testing were normal. DUSN is usually associated with segmental pigmentary changes and recurrent bouts of inflammation due to retinal damage from the offending nematode, but the pattern in our patient was equally progressive throughout the entire peripheral retina over 20 years. There are documented cases of women who are X-linked RP carriers having some pigmentary changes in the fundus due to the phenomenon of X inactivation. Those cases, however, would be expected to have segmental changes throughout the fundus of both eyes. Our patient manifested diffuse RP changes covering one entire retina while the contralateral eye remained completely normal.
In this case, the fundus findings, ERG results, and characteristic donut-shaped visual field loss made unilateral retinitis pigmentosa (RP) secondary to a somatic mutation a distinct possibility. It is known that most cases of suspected unilateral retinitis pigmentosa ultimately have another explanation. In order to make a reliable diagnosis of unilateral retinitis pigmentosa, it is necessary to rule out as best as possible all other explanations, confirm clinical signs of RP are truly unilateral, and follow the patient for an extended period (at least 5 years) to rule out the possibility of asymmetric inherited RP.
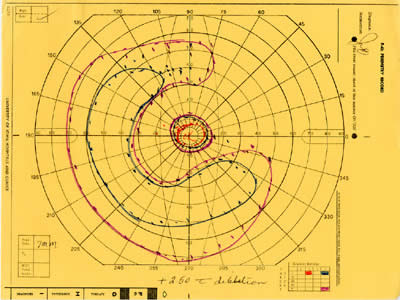
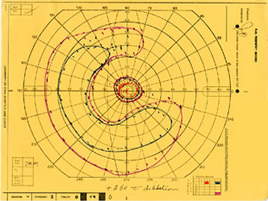
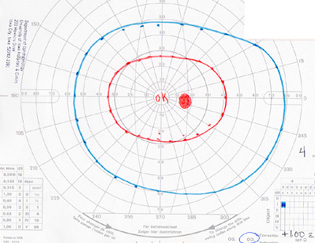
The patient was examined several times in the twenty years that followed. In the left eye, the peripheral visual field continued to deteriorate leaving only a small central island of preserved visual field (see Figure 5), while the visual field in the right eye remained normal (see Figure 6).
| Figure 5A: Visual field, OS, in 1985. Ring scotoma is evident | Figure 5B: Visual field, OS, in 1987 demonstrates further loss of peripheral visual field. | Figure 5C: Visual field, OS, in 1997 demonstrates complete loss of peripheral field with preserved central visual field. |
 |
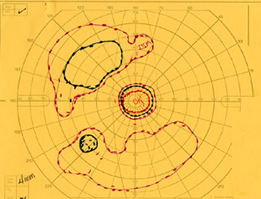 |
 |
| Figure 6: Visual field, OD, in 1997. All fields in the right eye remained completely normal over the course of care. |
 |
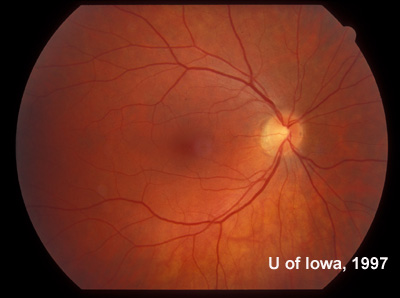
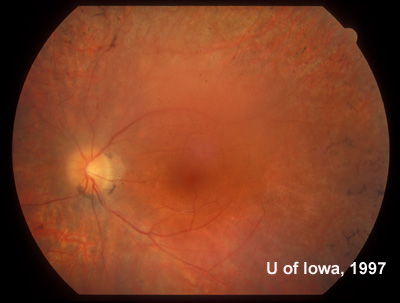
Fundus changes continued to progress in a unilateral manner, as well. In the left eye, progressive bone-spicule-like pigment changes developed throughout the peripheral retina. Centrally, the foveal reflex was lost and irregularity in the vitreoretinal interface developed. The disc became somewhat pale and retinal arteriolar narrowing became evident in the left eye. At no time was there ever segmental difference in the peripheral retinal changes. None of these changes occurred in the right eye (see Figures 7 and 8). In the left eye, a substantial posterior sclerotic cataract developed, while the crystalline lens in the contralateral eye remained clear. There was never any inflammation in either eye on all examinations. Repeat ERG continued to show an extinguished signal in the affected left eye and a normal reading in the right eye (see Figure 9).
| Figure 7A: Sixty degree view of the posterior fundus, right eye, more than twenty years after onset of symptoms in the left eye. | Figure 7B: Sixty degree view of the posterior fundus, left eye demonstrates arteriolar narrowing and retinal pigment atrophy and bone-spicule changes. A posterior sclerotic cataract contributes to the hazy view. |
 |
 |
| Figure 8A: Mid-peripheral view of the fundus in the right eye twenty years after the onset of symptoms in the contralateral eye. No abnormalities were seen. | Figure 8B: Mid-peripheral view of the fundus in the left eye (adjacent to the CHRPE first documented in 1975). RPE atrophy and bone-spicule changes are evident. The same bone-spicule changes were seen throughout the peripheral fundus of the left eye. |
 |
 |
| Figure 9A: Photopic ERG. Amplitude is normal in the right eye (OD) and extinguished in the left eye (OS). | Figure 9B: Scotopic ERG. Amplitude is normal in the right eye (OD) and extinguished in the left eye (OS). |
 |
 |
Several of the family members of this patient were also examined. None showed any retinal degeneration. Electroretinograms were performed on the patient's two sisters in their 30's and her 11-year-old son; the results were normal in all cases. At the time of the patient's most recent examination in July of 2005, all of the classic findings of retinitis pigmentosa persisted in the left eye only. The right eye remained completely normal.
Discussion: Retinitis pigmentosa (RP) is the term used to describe a group of degenerative, inherited disorders of the retina characterized by progressive photoreceptor damage and gradual atrophy and cell death of the photoreceptors and adjacent cell layers of the retina. Presenting symptoms of the disease include nyctalopia (night blindness), peripheral visual field loss, and sometimes loss of central visual acuity or visual field. The fundus of a patient with RP is characterized by mottling of the retinal pigment epithelium (RPE), followed by clumping of disrupted retinal pigment epithelium distributed in bone-spicule formations, attenuated retinal vessels, and waxy pallor of the optic disc. Autosomal recessive, autosomal dominant, and X-linked inheritance patterns of retinitis pigmentosa have all been clearly described and multiple gene defects associated with each inheritance pattern have been defined.
Unilateral retinitis pigmentosa is a very rare disease. The majority of suspected cases of unilateral retinitis pigmentosa ultimately are discovered to have another explanation (see differential diagnosis). Proper diagnosis of unilateral retinitis pigmentosa requires that all other explanations be ruled out and that the clinical and ERG findings of RP are well-defined in one eye and completely absent in the contralateral eye. These conditions must be true during extended follow-up (at least 5 years) to rule out the possibility of asymmetric inherited RP.
Unilateral RP is proposed to be the result of a somatic mutation during embryogenesis causing some percentage of cells within the body of the grown patient to carry a gene mutation with the potential to cause retinitis pigmentosa. If this mutation is present in the group of cells that become the retina and RPE, then the clinical presentation of RP will develop in that eye alone and the contralateral eye will remain normal. Of course, the same mutation with the potential to cause RP could be present in some part of the developing embryo destined to become skeletal muscle or bone, in which case the mutation would be absolutely silent. It is quite possible that this type of clinically silent mutation occurs relatively often; the dermatology literature includes many examples of biopsy-proven somatic mutations that manifest in discrete segments of the skin while the remaining skin surface remains normal. In the case of retinitis pigmentosa, a somatic mutation will result in an atypical or unilateral manifestation of the disease only when the disease-causing mutation occurs in a part of the embryo that is destined to become the eye.
Patients affected with this unusual condition are understandably curious about potential inheritance. One might think that because unilateral RP is a somatic mutation that the risk of an affected patient passing the condition on to their children should be zero. However, the clinician must remember that it is theoretically possible - though exceptionally rare - for an RP-causing mutation to occur early enough in embryogenesis to affect a large percentage of the patient's cell line, perhaps including the cells destined to become the germ line. The risk, then, of a patient with unilateral RP passing the mutation along to their children is probably very, very low but not necessarily zero.
There is no proven treatement for any form of retinitis pigmentosa yet. Many antioxidant, vitamin, and nutritional supplement therapies have been proposed, but none shown any reproducible benefit for patients with RP. At this point, treatment is supportive and includes low vision assistance, genetic testing, counseling, and treatment of related conditions if they develop (cataract or cystoid macular edema). An increasing number of specific gene mutations causing the condition have been discovered. Future developments in medicine and gene therapy may make early interventions possible for certain mutations in years to come.
EPIDEMIOLOGY
|
SIGNSIn the case of unilateral RP, the findings listed below are distinctly present in the affected eye and completely absent in the fellow eye:
|
SYMPTOMS
|
TREATMENT
|
Graff JM, Stone EM: Unilateral Retinitis Pigmentosa: Visual field changes in a 31-year-old female. EyeRounds.org. May 8, 2008; Available from: http://www.EyeRounds.org/cases/49-Unilateral-Retinitis-Pigmentosa.htm.

Ophthalmic Atlas Images by EyeRounds.org, The University of Iowa are licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License.

Address
University of IowaLegal
Related Links
