
Chief complaint: Eye crossing
History of Present Illness
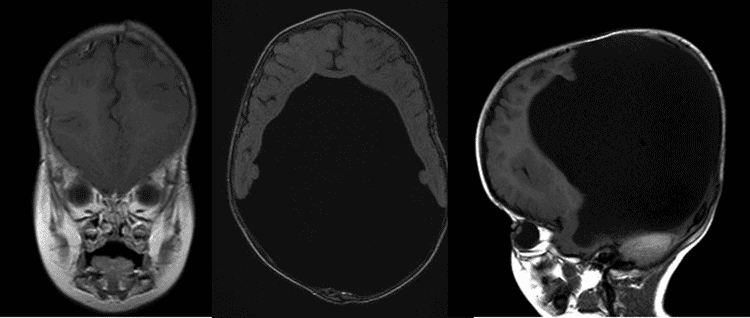
The patient is a 15 month-old female at presentation to the eye clinic, with history of severe hydrocephalus at birth. She was also diagnosed with alobar holoprosencephaly at birth with seizures. She was originally referred for eye crossing. We present her remarkable clinical course.
Past Medical History:
Past Surgical History:
Family History: No known family history of holoprosencephaly, amblyopia or strabismus.
Social History: Patient lives at home with parents and two sisters.
Medications: None
Visual Acuity: Central, unsteady and maintained OD and central, unsteady and maintained OS
Teller acuity testing:
Pupils: Equally round and briskly reactive, no relative afferent pupillary defect.
Stereo Vision: Unable to test
Motility and Strabismus:
Cycloplegic Refraction:
External Exam: Notable for large head circumference
Slit Lamp Exam: Normal anterior segment exam OU without evidence of cataracts or other media opacities.
Normal appearing optic nerves and normal dilated fundus examination. No sign of optic nerve hypoplasia in either eye.
 |
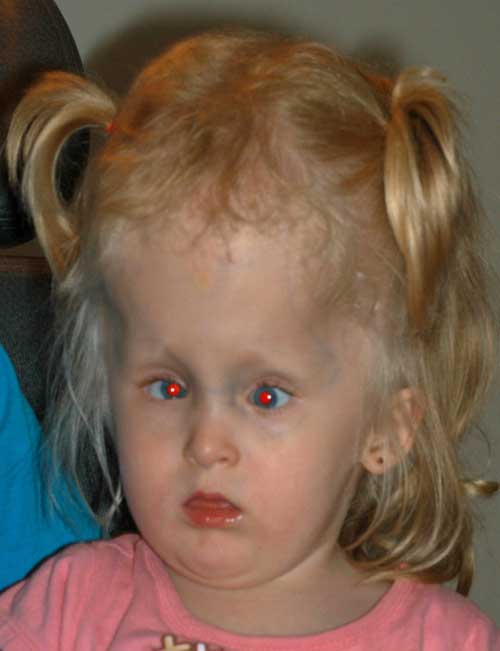
At this point, after discussion with her family, surgery for strabismus was deferred and correction of her hyperopia was attempted. Glasses were prescribed. She was not able to wear glasses comfortably and contact lenses were tried. Patching was performed to treat her amblyopia. The patient was followed and at age 3, her parents wanted to proceed with strabismus surgery to “uncross” her eyes.
Visual Acuity: Central, CUSM OD and CUSM OS
Motility and Strabismus:
 |
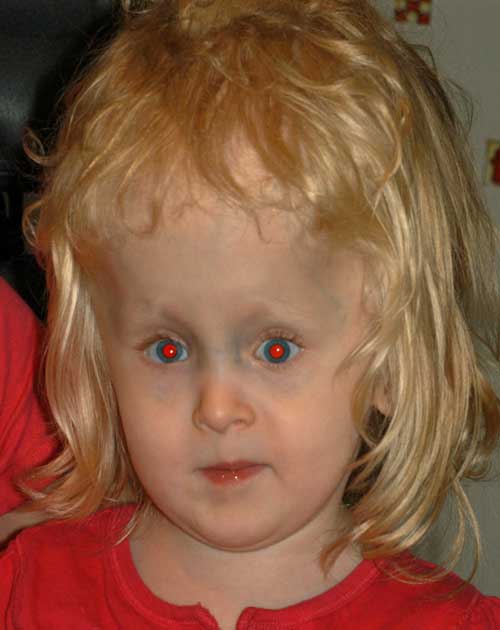
At the time of strabismus surgery, intraoperative forced ductions showed restrictions of both medial rectus muscles and the patient was found to have an anomalous insertion of the medial rectus muscle. The medial rectus muscle insertion prior to disinsertion was found 7 mm from limbus (more posterior than the expected 5.5 mm from the limbus). Secondary to the anatomy not being normal, a conservative approach was performed and she underwent bilateral medial rectus recessions of 5.5 mm, leaving the medial rectus at 12.5 mm from the limbus.
Post-operatively, she had a small residual variable esotropia with small vertical deviation and a more noticeable elevation deficiency of the left eye.
 |
Her parents were happy with alignment; however, with time, she developed a noticeable, right hypertropia. At age 6, her family decided to proceed with a second strabismus surgery to address the vertical misalignment.
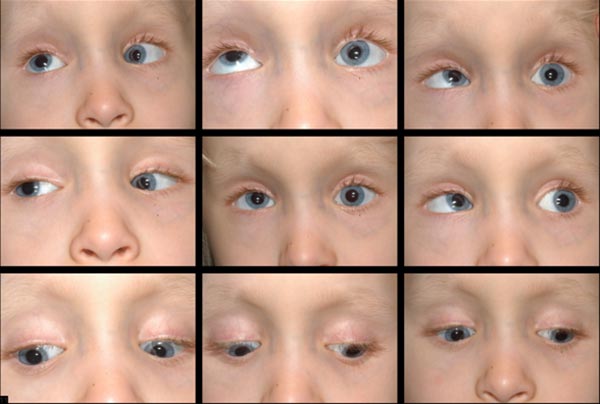 |
A left inferior rectus recession vs a right superior rectus recession was planned, with decision regarding which procedure would be performed based on intraoperative forced ductions. Intra-operative forced ductions showed a tight left inferior rectus, which is consistent with the monocular elevation deficiency we had previously considered. A left inferior rectus recession of 6 mm was performed, moving the muscle from 8 mm posterior to the limbus, where it was found, to 14 mm posterior to the limbus.
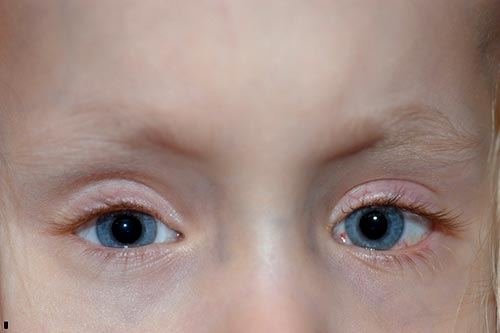 |
A few months after her second surgery, her mother began to notice that her right eye drifted upwards in times of inattention. She was seen in clinic and noted to have a manifest dissociated vertical deviation on the right. It was decided to proceed again with strabismus surgery and an 8 mm right superior rectus recession by hangback method, moving the muscle from 8 mm posterior to the limbus, where it was found, to 16 mm posterior to the limbus.
The patient did well post-operatively (Figure 6), and has been progressively developing her visual skills. She has done remarkably well overall and is walking and now reading.
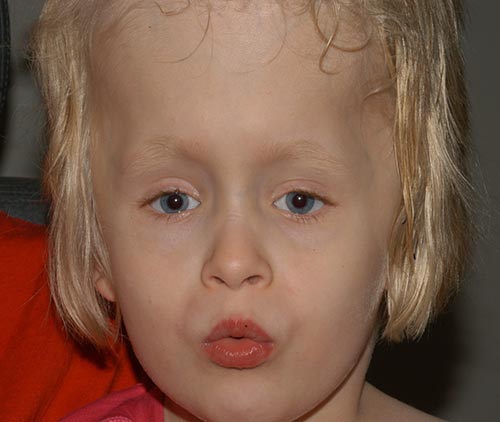 |
Holoprosencephaly with infantile esotropia and complex strabismus, with variability in medial rectus muscle insertions.
Holoprosencephaly is a type of cephalic disorder characterized by the failure of the prosencephalon (the embryonic forebrain) to develop, leading to a single-lobed brain structure and severe skull and facial defects. There are three classifications of holoprosencephaly: alobar holoprosencephaly, semi-lobar holoprosencephaly, and lobar holoprosencephaly.
Alobar holoprosencephaly accounts for two thirds of affected patients, and is the most serious form, characterized by failure of the brain to separate into two halves. This results in a single primitive ventricle, absent olfactory bulbs and optic tracts and severe developmental abnormalities. It is usually associated with severe facial anomalies, including closely spaced eyes, small head size, cleft lip and palate. Semi-lobar holoprosencephaly, accounting for a quarter of holoprosencephaly cases, is an intermediate form of the disease and is characterized by partially separated cerebral hemispheres and a single ventricle. Lobar holoprosencephaly is the least severe form, in which the patient's brain may be nearly normal; there is a distinct fissure between developed central lobes, and some fusion of the brain structures is present (Nanni, 2000). In most cases of holoprosencephaly, brain malformations are incompatible with life. In less severe cases, babies are born with normal or near-normal brain development and varying degrees of facial deformity.
Midline craniofacial defects are the hallmark of holoprosencephaly and may include microcephaly, hypotelorism (abnormally closely spaced eyes), nasal abnormalities, such as nasal flattening or a single naris, and the upper lip and palate defects such as cleft palate or a single front incisor. Cyclopia may be present in the most severe forms where a nose-like proboscis is present over a single eye in the middle of the face (Nanni, 2000). The degree of facial deformity is thought to indicate the severity of intracranial defects. Associated co-morbidities include dysfunction of the pituitary gland and hypothalamus, resulting in body temperature dysregulation, seizures, and mental retardation of varying severity (Dubourg, 2007). Hypotonia and dystonia have also been observed (Barkovich, 2002).
Holoprosencephaly occurs during the first weeks of intrauterine life. Prevalence of holoprosencephaly in early embryonic development is 1:250, decreasing to 1:10,000-1:20,000 at term (Nanni, 2000). There is no known cause of holoprosencephaly, although there have been many suggested risk factors, including maternal diabetes (1% risk, 200-fold increase) (Barra, 1983), infections during pregnancy, such as the TORCH infections (Munke, 1989), and exposure to toxic substances, including alcohol, lithium, Thorazine, hormones, anticonvulsants and retinoic acid (Nanni, 2000). Most cases are considered to occur sporadically, although holoprosencephaly has been found to have a genetic basis as well. Familial holoprosencephaly has been seen inherited in both autosomal dominant and autosomal recessive patterns. Chromosomal anomalies have also been associated with holoprosencephaly, with trisomy 13 being the most common, although this is not a constant association (Kallen, 1992).
Pathogenesis is thought to involve a defect in the signaling genes responsible for regulating neural tube patterning. Intracranial findings include varying cortical hypoplasia, varying fusion of the diencephalon, basal ganglia and thalamus and presence of a dorsal cyst (arising from fused thalami) expanding from a partially blocked 3rd ventricle (Simon, 2001). Hydrocephalus, caused by abnormal accumulation of CSF in the ventricles, is not uncommon in holoprosencephaly and is thought to be due to malformation of the ventricles or excessive CSF production. This often complicates holoprosencephaly classification, as the brain is compressed and the previously microcephalic cranium is allowed to expand prior to fusion of the cranial sutures (Tripathi, 2009). It is important to address the patient’s vision to allow for optimal interaction with his or her surrounding environment. Often glasses are not tolerated due to the degree of facial asymmetry and structural abnormalities present. In these situations, we feel that contact lenses should be considered as a way to improve visual function.
Although there is variation of medial rectus muscle insertion in the general population, this case is remarkable for the anomalous extraocular muscle insertions, specifically the 7 mm distance of the medial recti from the limbus. As mentioned above, midline defects are common in holoprosencephaly, which may explain why the medial recti were preferentially involved. With the exception of cyclopia, little has been published on ocular and strabismic associations with holoprosencephaly.
Overall, treatment is highly individualized based on the patient’s severity and configuration of malformation. Treatment is supportive and symptomatic and prognosis depends greatly on the type of holoprosencephaly and its associated anomalies (Nanni, 2000).
In the case presented above, one of the most significant considerations for the patient's family was how to intervene to improve vision and allow for continued growth and development. The patient was critically ill in the first few years of life, and palliative care was initially discussed as a viable option. The patient's family wanted to continue treatment and seek out possible interventions to improve quality of life. When faced with challenging situations such as this, it is essential for health care providers to assist families to make decisions in a respectful environment without judgment or undue influence. The American College of Critical Care Medicine Task Force published clinical practice guidelines addressing support of the family in the patient-centered intensive care unit (Davidson, 2007). Through good communication, conflict management and meeting facilitation skills, families can be involved in a shared decision-making model in which families are not solely responsible for all medical decisions autonomously, nor are medical providers providing paternalistic care. During family meetings, it is recommended that family members be asked open-ended questions about their understanding of the patient’s care, their fears and coping strategies. Care-providers are then encouraged to repeat the family’s sentiments to allow for the development of trust in the team and the decision-making process. After this, the practitioners should provide clear and honest information in accessible language, with the opportunity to ask questions. The goal of the discussion is consensus, which is helped by respectful acknowledgment of all opinions.
Penticuff and Arheart studied the effectiveness of meetings between healthcare providers and parents in a neonatal intensive care unit setting, and showed that shared decision-making resulted in fewer conflicts, unrealistic parental expectations, and improved collaboration, as well as helping parents better understand their child’s medical situation (Penticuff, 2005). Family stress levels are shown be decreased with open and effective communication, as well as an environment of hope (Davidson, 2007). Importantly, such patient-centered care has been shown to improve clinical outcomes, as well (Lewin, 2001).
Summary
|
Signs
|
Symptoms
|
Treatment
|
Barkovich AJ, Simon EM, Clegg NJ, Kinsman SL, Hahn JS. Analysis of the cerebral cortex in holoprosencephaly with attention to the sylvian fissures. AJNR Am J Neuroradiol. 2002;23(1):143-50.
Blaas HG, Eriksson AG, Salvesen KA, Isaksen CV, Christensen B, Møllerløkken G, Eik-Nes SH. Brains and faces in holoprosencephaly: pre- and postnatal description of 30 cases. Ultrasound Obstet Gynecol. 2002;19(1):24-38.
Barra Jr M, Hanson JW, Currey K, Sharp S, Toriello H, Schmickel RD, Wilson GA. Holoprosencephaly in infants of diabetic mothers. J Pediatr 1983; 102: 565D8.
Davidson JE, Powers K, Hedayat KM, Tieszen M, Kon AA, Shepard E, Spuhler V, Todres ID, Levy M, Barr J, Ghandi R, Hirsch G, Armstrong D. Clinical practice guidelines for support of the family in the patient-centered intensive care unit: American College of Critical Care Medicine Task Force 2004-2005. American College of Critical Care Medicine Task Force 2004-2005, Society of Critical Care Medicine. Crit Care Med. 2007;35(2):605-22.
Dubourg C, Bendavid C, Pasquier L, Henry C, Odent S, David V. Holoprosencephaly. Orphanet J Rare Dis. 2007 2;2:8.
Kallen B, Castilla EE, Lancaster PAL, et al. The cyclops and the mermaid: an epidemiological study of two types of rare malformation. J Med Genet 1992;29:30–35.
Lewin SA, Skea ZC, Entwistle V, Zwarenstein M, Dick J. Interventions for providers to promote a patient-centred approach in clinical consultations. Cochrane Database Syst Rev. 2001;(4): CD003267. (PMID:11687181)
Munke M. Clinical, cytogenetic and molecular approaches to the genetic heterogeneity of holoprosencephaly. Am J Med Genet 1989;34:237–245.
Nanni L, Schelper RL, Muenke MT. Molecular genetics of holoprosencephaly. Front Biosci. 2000 1;5:D334-42.
Penticuff JH, Arheart KL.Effectiveness of an intervention to improve parent-professional collaboration in neonatal intensive care. J Perinat Neonatal Nurs. 2005;19(2):187-202.
Scott WE, Jackson OB. Double elevator palsy: the significance of inferior rectus restriction. Am Orthopt J. 1977;27:5-10.
Simon EM, Hevner RF, Pinter J, Clegg NJ, Delgado M, Kinsman SL, Hahn JS, Barkovich AJ. The dorsal cyst in holoprosencephaly and the role of the thalamus in its formation. Neuroradiology. 2001;43(9):787-91.
Tripathi AK, Agrawal D, Sedain G. Hydrocephalic holoprosencephaly: An oxymoron? Insights into etiology and management. J Pediatr Neurosci. 2009;4(1):41-3.
Consent for using photos and video obtained from patient’s mother.
Kemp PS, Casey, G, Longmuir SQ. Holoprosencephaly and Strabismus.Eyerounds.org. June 12, 2012, Available from: https://eyerounds.org/cases/151-holoprosencephaly-strabismus.htm

Ophthalmic Atlas Images by EyeRounds.org, The University of Iowa are licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License.

Address
University of IowaLegal
Related Links
