
Right eye pain and decreased vision
The patient is a 34-year-old female with a history of Crohn’s disease with inflammatory arthritis who was referred to the UIHC neuro-ophthalmology clinic for evaluation of painful vision loss in her right eye. She first developed pain in her right eye four weeks prior to presentation. The pain was worse with eye movement. Over the next couple of days, the eye pain was followed by progressive vision loss that she described as a “comma shaped dark/blurry spot” in the middle of her vision. She saw an outside ophthalmologist, who noted Visual Acuity (VA) of 20/40 in the right eye (OD), VA of 20/25 in left eye (OS), and a swollen right optic nerve on dilated fundus exam. She was subsequently referred to UIHC neuro-ophthalmology for further evaluation. Upon presentation to UIHC, her vision had stabilized, but had not improved. She continued to have pain.
She denied prior similar episodes, diplopia, or oscillopsia. She had a history of migraines but denied a change in frequency, duration, or character. She denied ataxia, paresthesias, fevers, sweats, or rigors.
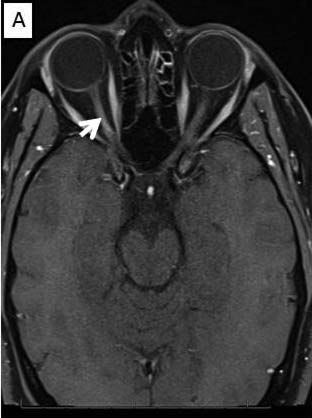
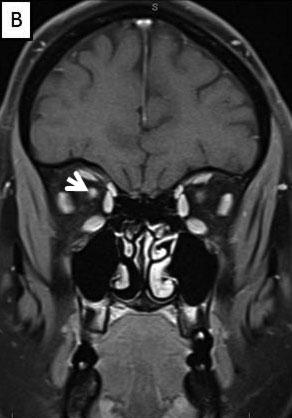
Magnetic Resonance Imaging (MRI) brain/orbits with and without contrast (Figure 3):
 |
 |
 |
 |
 |
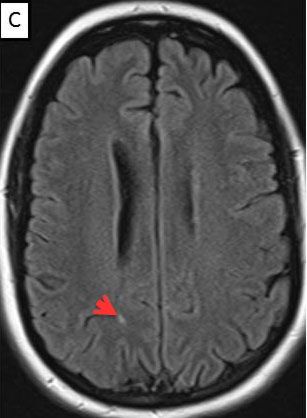 |
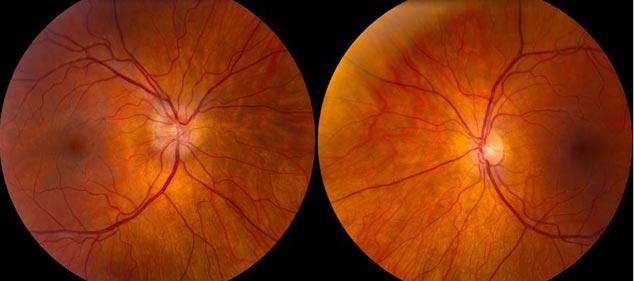
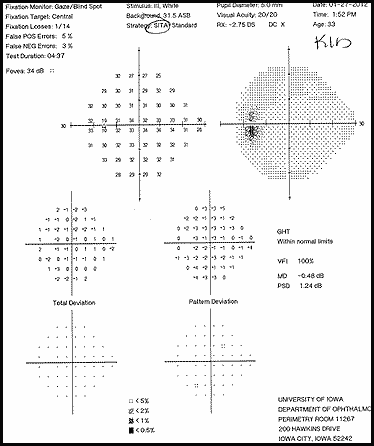
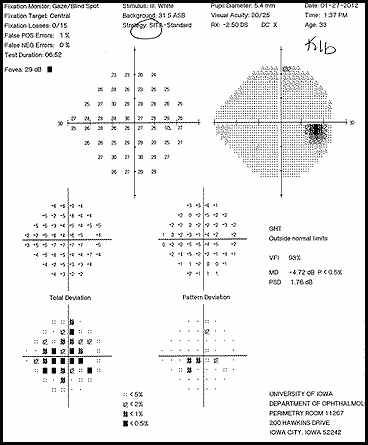
The patient had a subacute-onset of painful monocular vision loss OD. Examination findings were consistent with a right optic neuropathy. Critical flicker fusion was moderately decreased OD. HVF revealed a central scotoma OD. OCT of the optic nerves revealed increased RNFL thickness OD. Laboratory work-up was unremarkable, except for mildly elevated inflammatory markers (ESR 39 mm/hr and CRP 1.2 mg/dL). Finally, MRI brain/orbits with and without contrast revealed diffuse enhancement of the retrobulbar portion of the right optic nerve and a single subcortical white matter lesion in the posterior right hemisphere of uncertain significance. The clinical evidence was most in favor of a diagnosis of optic neuritis.
The patient was treated in accordance with the Optic Neuritis Treatment Trial.[1] The patient received IV methylprednisolone (1g x 3d), followed by oral prednisone (1mg/kg x 11d), and then tapered off of steroids. A complete discussion of optic neuritis can be found at http://www.EyeRounds.org/cases/159-optic-neuritis.htm. As expected, the patient showed significant improvement at her six week follow-up. The pain had resolved and her visual fields were full OU with a residual 0.3 log unit RAPD OD. Her critical flicker fusion also showed significant improvement (25.1 Hz, standard deviation 2.0). At this six week visit, cessation of the TNF-α antagonist (adalimumab) therapy was recommended, since demyelinating diseases such as optic neuritis and MS have been associated with anti-TNF-α therapy (see discussion below). Furthermore, her family history of a mother with MS may have predisposed her to demyelinating disease while taking an anti-TNF-α agent. However, the patient was very reluctant to switch from the anti-TNF-α therapy because of the relief she had received from her Crohn’s disease and inflammatory arthritis while on this therapy. Therefore, we consulted her gastroenterologist to discuss alternative therapy options with her. Upon meeting with her gastroenterologist, it was decided that the risk of demyelinating disease outweighed that of the therapeutic benefit of adalimumab, and she was switched to sulfasalazine.
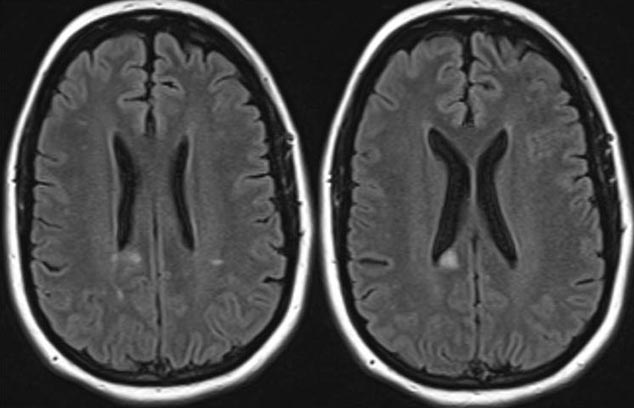
At her three-month follow-up appointment, the patient had obtained a full visual recovery from her attack of optic neuritis OD. However, after switching off of adalimumab therapy, she was experiencing worsening of her Crohn’s disease and increased arthralgias, especially in her large joints. Furthermore, at this visit she complained of numbness in her right lower extremity. A repeat MRI brain with contrast showed several new periventricular white matter lesions, with enhancement, consistent with active demyelination (Figure 4). Neurology was consulted and further work-up, including lumbar puncture, was recommended before any disease-modifying therapy for demyelinating disease was initiated.
 |
In recent years, tumor necrosis factor alpha (TNF-α) antagonists have revolutionized the treatment of autoimmune inflammatory diseases, such as Crohn’s disease, rheumatoid arthritis, and ankylosing spondylitis. Although this method of therapy has provided significant benefits to patients with inflammatory autoimmune diseases, as demonstrated in our case, there has been a growing body of literature that describes various adverse effects associated with anti-TNF-α therapy. These side effects include systemic lupus erythematosus, malignant lymphoma, severe infections, and demyelinating diseases.[2] Of particular interest in the field of ophthalmology is the association of anti-TNF-α agents with demyelinating diseases. The variety of clinical presentations that could be encountered in an ophthalmology clinic might include optic neuritis, transverse myelitis, Guillain-Barre syndrome (GBS), and MS.[3] The remainder of the discussion will focus on a literature review of the association of anti-TNF-α therapy and demyelinating diseases.
Initial interest in the association between TNF-α and demyelinating disease was first generated when TNF-α was demonstrated to be capable of causing gradual demyelination of murine nerve tissue cultures in vitro.[4] The levels of TNF-α were subsequently shown to be elevated in patients with chronic progressive MS compared to patients with stable MS.[5] Therefore, it was concluded that the level of TNF-α correlated with the severity and progression of the disease. Next, in vitro studies demonstrated that TNF-α was cytotoxic to oligodendrocytes, which are the cells selectively damaged in MS.[6] This suggested that the elevated levels of TNF-α may actually be central to the MS disease process. With TNF-α possibly playing a key role in MS pathology, the therapeutic role of anti-TNF-α agents was next tested in a mouse model that replicated that of MS.[7] Results from this mouse study showed that anti-TNF-α agents were successful in preventing autoimmune demyelination, suggesting that anti-TNF-α agents could be utilized to prevent and/or treat human demyelinating diseases such as MS.
Because anti-TNF-α agents improved a mouse model of MS, an open-label phase I safety trial was performed in which two patients with rapidly-progressive MS were treated with IV infusions of a humanized mouse monoclonal anti-TNF antibody.[8] Results from this study were quite concerning as the therapy was not effective in treating MS. On the contrary, the treatments corresponded with immune activation and increase in disease activity. Similar results were obtained during a phase II study, in which anti-TNF-α therapy (Lenercept) resulted in increased MS attack frequency, duration, and severity.[9] Therefore, it has been concluded that anti-TNF-α agents are not appropriate for treating MS and that patients treated with such agents should be monitored closely for the development of demyelinating disease.
Since these initial studies, case reports have more clearly described the adverse effects of anti-TNF-α agents. There are a variety of manifestations of demyelinating disorders associated with anti-TNF-α therapy, which include, in descending order, optic neuritis, MS, demyelinating neuropathy, GBS, and transverse myelitis, all of which have a fairly low incidence.[10] Currently, it is believed that patients will rarely experience demyelinating disease within the first several months of initiating therapy. The average time from the introduction of therapy to the onset of demyelinating disease is 5 months.[3] The current recommendation for treatment of anti-TNF-α associated demyelination disease is to immediately discontinue the drug with the development of any neurological symptoms (parasthesia, change in vision, etc.), even if the initial symptoms are mild, transient, or resolve without treatment.[3] Continuation of anti-TNF-α therapy after development of neurologic symptoms can be associated with a poor outcome and permanent neurologic deficits.[11] Restarting the anti-TNF-α agent after experiencing adverse neurologic symptoms is associated with a high recurrence rate and a high incidence of permanent deficits. Prompt cessation of anti-TNF-α therapy upon initial appearance of neurologic symptoms most often showed partial or complete recovery of neurologic deficits.[3] However, a small subset of patients experience persistent neurologic deficits.[12] In the case of continued neurologic symptoms, initiation of MS disease-modifying therapy such as glatiramer acetate, IFN β-1a, or IFN β-1b may be considered, as these drugs have shown to reduce the number of MS relapses and possibly long-term disability.[13] It has been recommended that anti-TNF-α therapy should be avoided in patients with a preexisting MS diagnosis, a history of unexplained central neurologic signs, or a family history of MS.[14]
Optic neuritis in the context of anti-TNF-α therapy
Differential DiagnosisOptic neuritis
Neuromyelitis optica |
SignsVisual field abnormalities
Visual acuity abnormalities
Pupillary abnormalities
|
Symptoms
|
Treatment
|
In the case above, the patient experienced significant relief from her Crohn’s disease and inflammatory arthritis with anti-TNF-α therapy. The success of relieving symptoms in this patient resulted in her reluctance to cease therapy with adalimumab, despite being warned of the potential adverse effects. Several key learning points can be taken from this case. Clinicians must be cognoscente of the association of anti-TNF-α therapy with demyelinating disorders. Some of the current literature would have argued that this patient, who had a mother with MS, should have never been started on anti-TNF-α therapy due to her increased predisposition to demyelinating disorders. Finally, although this patient was experiencing significant relief from her Crohn’s disease and inflammatory arthritis, the anti-TNF-α therapy should be discontinued at the first sign of neurologic symptoms despite the transient nature of the initial symptoms and resolution with treatment.
1. Optic Neuritis Study Group. Visual function 15 years after optic neuritis: a final follow-up report from the Optic Neuritis Treatment Trial. Ophthalmology. 2008; 15:1079-1082.
2. Scheinfeld N. A comprehensive review and evaluation of the side effects of the tumor necrosis factor alpha blockers etanercept, infliximab and adalimumab. J Dermatolog Treat. 2004; 15:280-294.
3. Mohan N, Edwards ET, Cupps TR, Oliverio PJ, Sandberg G, et al. Demyelination occurring during anti-tumor necrosis factor alpha therapy for inflammatory arthritides. Arthritis Rheum. 2001; 44:2862-2869.
4. Selmaj K, Raine CS. Tumour necrosis factor mediates myelin and oligodendrocyte damage in vitro. Ann Neurol. 1988; 23:339-346.
5. Sharief MK, Hentges R. Association between tumor necrosis factor-alpha and disease progression in patients with multiple sclerosis. N Engl J Med. 1991; 325:467-472.
6. Selmaj K, Raine CS, Farooq M, Norton WT, Brosnan CF. Cytokine cytotoxicity against oligodendrocytes. Apoptosis induced by lymphotoxin. J Immunol. 1991; 147:1522-1529.
7. Selmaj KW, Raine CS. Experimental autoimmune encephalomyelitis: immunotherapy with anti-tumor necrosis factor antibodies and soluble tumor necrosis factor receptors. Neurology. 1995; 45:44-49.
8. van Oosten BW, Barkhof F, Truyen L, Boringa JB, Bertelsmann FW, et al. Increased MRI activity and immune activation in two multiple sclerosis patients treated with the monoclonal anti-tumor necrosis factor antibody cA2. Neurology. 1996; 47:1531-1534.
9. The Lenercept Multiple Sclerosis Study Group and The University of British Columbia MS/MRI Analysis Group. TNF neutralization in MS: results of a randomized, placebo-controlled multicenter study. Neurology.1999; 53:457-465.
10. Fernandez-Espartero MC, et al.-Demyelinating disease in patients treated with TNF antagonists in rheumatology: data from BIOBADASER, a pharmacovigilance database, and a systematic review. Semin Arthritis Rheum. 2011; 40:330-337.
11. Lozeron P, Denier C, Lacroix C, Adams D. Long-term course of demyelinating neuropathies occurring during tumor necrosis factor-alpha-blocker therapy. Arch Neurol. 2009; 66:490-497.
12. Seror R, Richez C, Sordet C, Rist S, Gossec L, et al. Pattern of demyelination occurring during anti-TNF-α therapy: a French national survey. Rheumatology (Oxford). 2013; 52:868-74.
13. Goldberg LD, Edwards NC, Fincher C, Doan QV, Al-Sabbagh A, et al. Comparing the cost-effectiveness of disease-modifying drugs for the first-line treatment of relapsing-remitting multiple sclerosis. J Manag Care Pharm. 2009; 15:543-55.
14. Toussirot E, Pertuiset E, Martin A, Melac-Ducamp S, Alcalay M, et al. Association of rheumatoid arthritis with multiple sclerosis: report of 14 cases and discussion of its significance. J Rheumatol. 2006; 33:1027-8.
Baldwin A, Thurtell MJ. Tumor Necrosis Factor Alpha Antagonist Precipitating Demyelinating Disease. EyeRounds.org. June 3, 2013; Available from: http://www.EyeRounds.org/cases/170-TNF-alpha-demyelinating.htm

Ophthalmic Atlas Images by EyeRounds.org, The University of Iowa are licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License.

Address
University of IowaLegal
Related Links
