
Chief Complaint: Mild decreased vision
History of Present Illness: A 57-year-old Caucasian male was referred to the University of Iowa Retina Service for retinal thickening and subretinal fluid in the right eye (OD) greater than the left eye (OS). The patient had noted a stable gray spot and visual distortion OD for the previous several weeks. He had stable chronic floaters but no flashes of light, eye pain, or other ocular symptoms.
Past Ocular History: OD injury at 15 years old
Past Medical History: Traumatic amputation of left index finger, premature ventricular contractions, hernia repair
Medications: None
Allergies: None
Family History: Mother with macular degeneration
Social History: Dairy farmer; no tobacco, alcohol, or drug use
Review of Systems: Negative
| OD | OS | |
|---|---|---|
| VA without correction | 20/80 | 20/40 +1 |
| VA with pinholes, without correction | 20/40 +1 | 20/25 +1 |
| Pupil examination | Brisk, consensual 3mm to 2mm; no relative afferent pupil defect | Brisk, consensual 3mm to 2mm; no relative afferent pupil defect |
| Visual Fields | Full by confrontation | Full by confrontation |
| Motility | Full | Full |
| Intraocular Pressure | 15 mm Hg by Tonopen | 13 mm Hg by Tonopen |
| External and slit lamp examination | Normal, cornea clear, AC deep and quiet, lens clear | Normal, cornea clear, AC deep and quiet, lens clear |
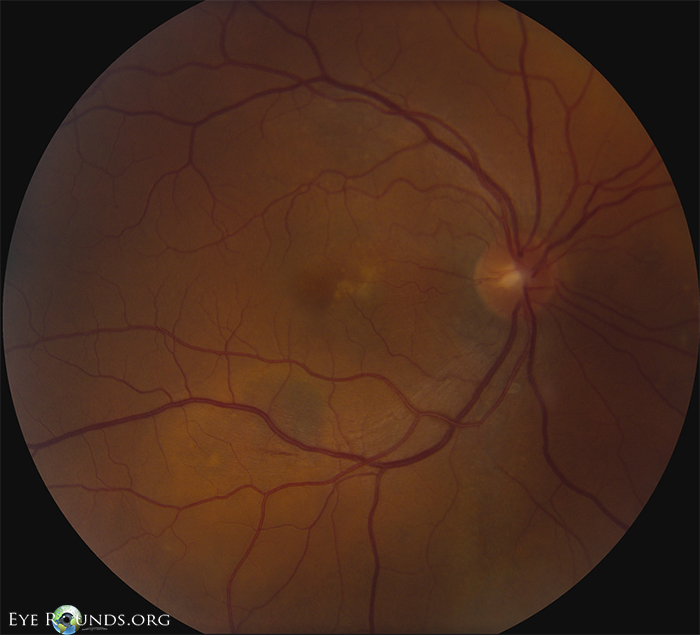
| Dilated fundus exam | Posterior vitreous detachment, normal disc, C/D 0.3, shallow multifocal pigment epithelial detachments (PEDs) between the nerve and fovea with adjacent subretinal fluid (SRF), scattered pigmentary changes at the fovea (See Figures 1, 3, 5 below) |
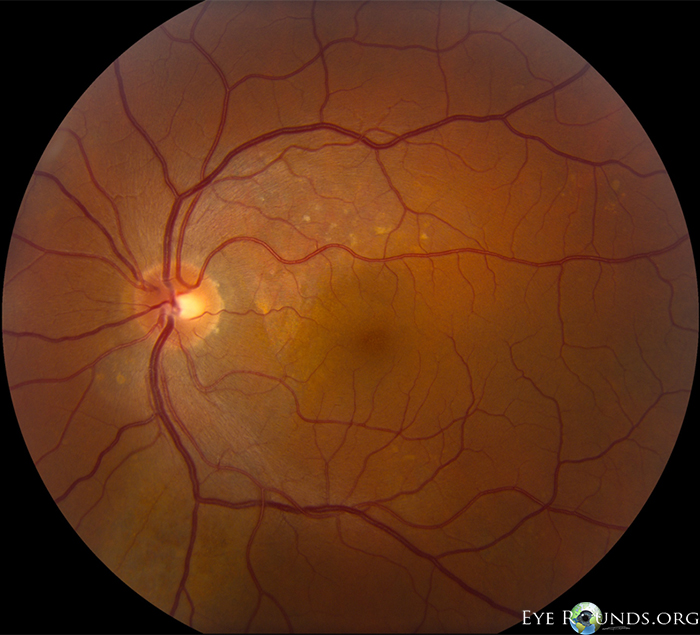
Posterior vitreous detachment, normal disc, C/D 0.3, thin subretinal fluid adjacent to nerve (~1DD), few small ill-defined pigmentary changes superonasal to the fovea (See Figures 2, 4, 6 below) |
| Figure 1: OD color fundus photograph at initial presentation showing a shallow PED nasal to fovea, SRF inferonasal to macula, and pigment clumping along inferotemporal arcade. | Figure 2: OS color fundus photograph at initial presentation showing SRF temporal to the disc and scattered yellow pigment superonasal to macula. |
|---|---|
 |
 |
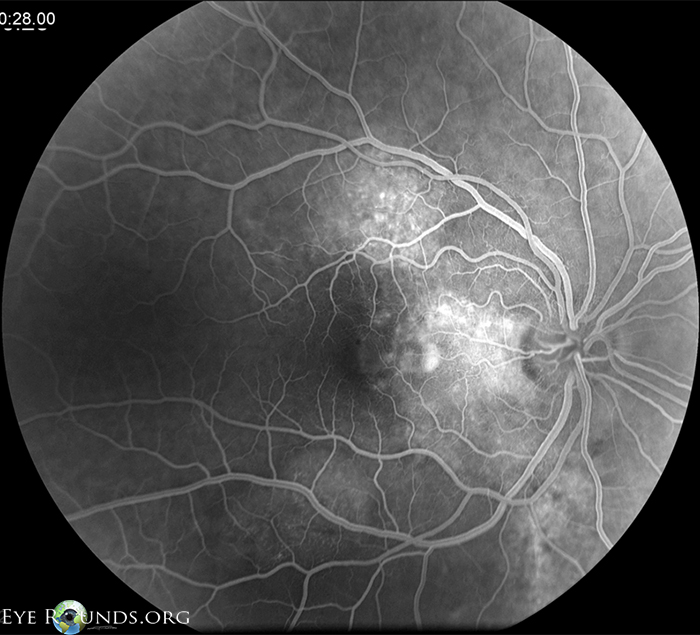
| Figure 3 : OD fluorescein angiography, late frames, at initial presentation showing patchy areas of leakage in the nasal macula and along the superior and inferior arcades. | Figure 4: OS fluorescein angiography, late frames, at initial presentation showing diffuse leakage in the superonasal macula. |
|---|---|
 |
 |
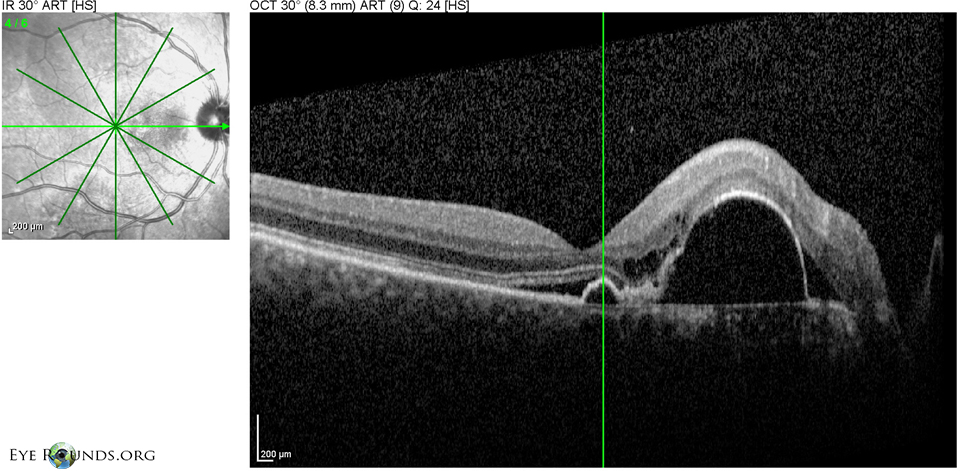
Figure 5: OD Spectralis OCT of the macula at initial presentation showing multiple PEDs with shallow SRF and mild overlying intraretinal cystic degeneration.
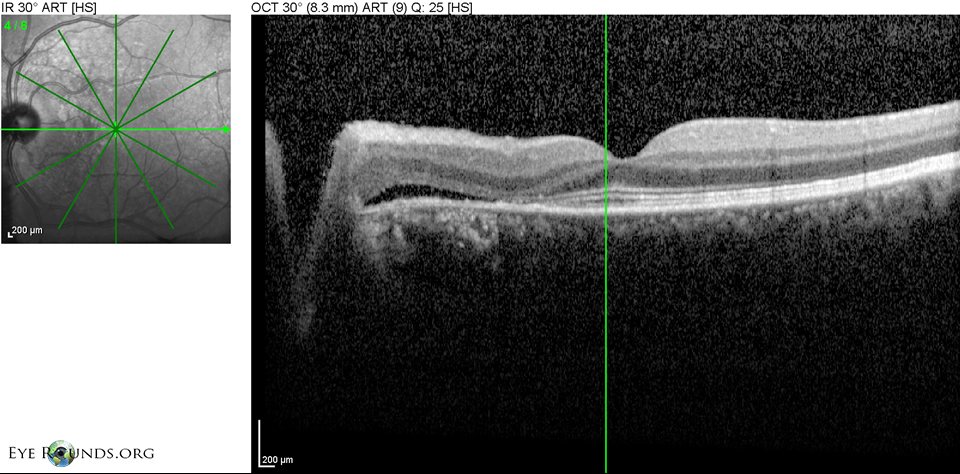
Figure 6: OS Spectralis OCT of the macula at initial presentation with shallow SRF temporal to the disc

Figure 7: OD Spectralis OCT of the macula at 10 month follow-up showing a large PED between the fovea and disc, with mild overlying intraretinal cystic degeneration.
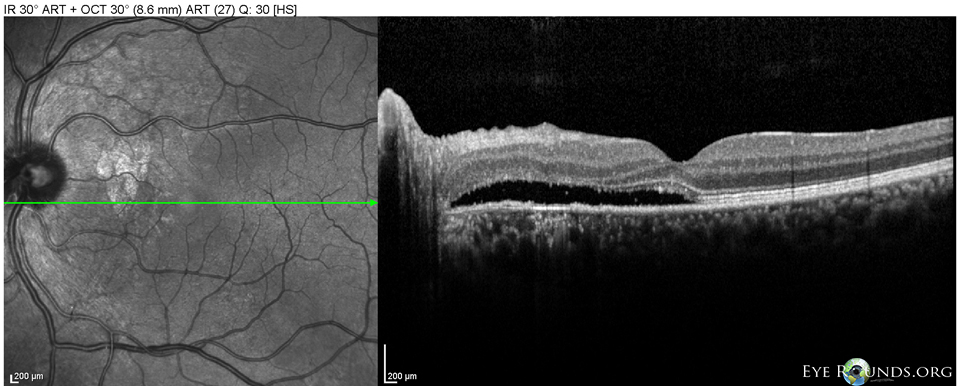
Figure 8: OS Spectralis OCT of the macula at 10 month follow-up showing worse shallow SRF between the nerve and fovea.
Due to the presence of multifocal serous pigmented epithelium detachments (PEDs) with adjacent subretinal fluid (SRF) OD and SRF OS, along with leakage on fundus fluorescein angiography (FFA), an initial diagnosis of central serous retinopathy (CSR) was made. The patient was started on oral ketoconazole 200 mg by mouth twice daily for three months but did not show improvement in symptoms or visual acuity.
At his ten month follow-up appointment, optical coherence tomography (OCT) showed minimal cystic changes in the macula, minimal SRF and PEDs OD and SRF OS (Figures 7, 8). FFA showed patchy areas of hyperfluorescence around the macula OU and the patient was started on oral eplerenone 25 mg twice daily for presumed CSR.
Follow-up examination at one year after initial presentation showed no improvement on OCT of his macula and therefore oral eplerenone was switched to oral spironolactone 50 mg daily with mild improvements in central macular thickness (CMT) and SRF OU. His visual acuity improved to 20/20 in both eyes with updated refraction. However, this improvement was accompanied by intolerable breast tenderness and the spironolactone was stopped.
Follow-up examination 3 months later showed decreased vision and recurrence of intraretinal cysts with increased CMT and SRF OU. At this time indocyanine green angiography (ICG) was obtained along with FFA. The ICGs revealed polypoidal lesions temporal to the disc in both eyes. (Figures 9, 10). The diagnosis of polypoidal choroidal vasculopathy (PCV) was made. It was felt that his previous improvement on spironolactone had been coincidental.
Intravitreal bevacizumab injections were given in both eyes with little improvement in vision or subretinal fluid. Subsequently, he was given three monthly intravitreal aflibercept injections in both eyes, with some improvement in subretinal fluid. Vision improved to 20/20 -1 OD and 20/20 OS from 20/25 +2 OD and 20/15 -2 OS since the initiation of intravitreal injections.
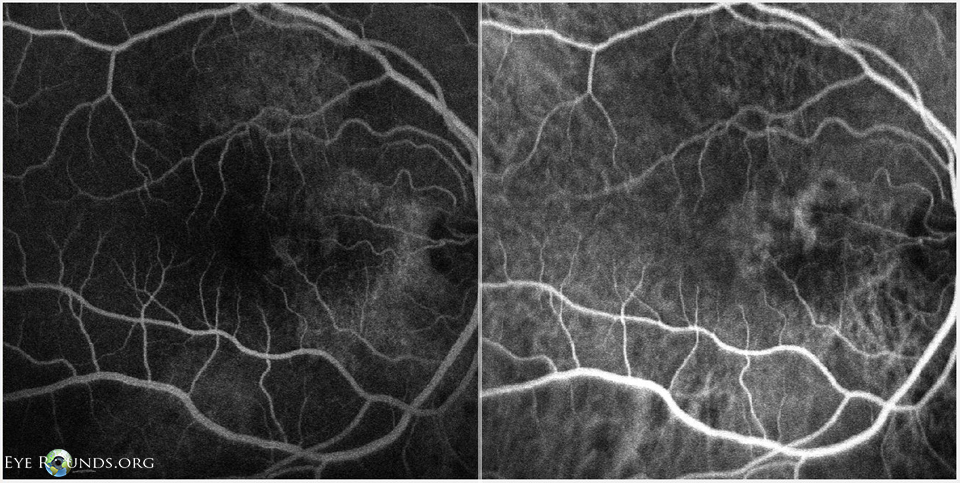
Figure 9: OD FFA (left) and ICG (right) showing punctate fluorescent lesions temporal to the optic nerve in the nasal macula, most consistent with polypoidal lesions.
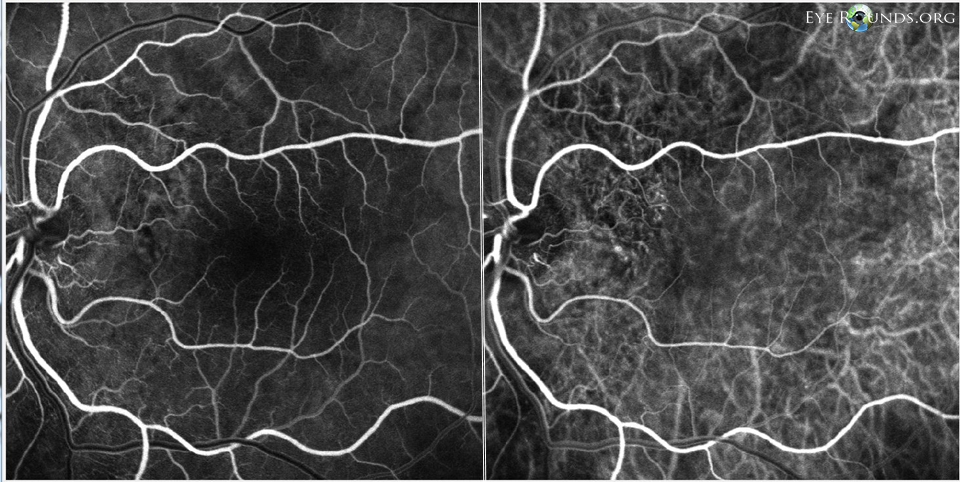
Figure 10: OS FFA (left) and ICG (right) showing subtle punctate areas of hyperfluorescence just temporal to the nerve, most consistent with polypoidal lesions.
Polypoidal choroidal vasculopathy (PCV) was first described in 1982 by Yannuzzi[1] and was further characterized in 1990 by Yannuzzi[8] and Kleiner[9], who referred to this disease as "posterior uveal bleeding syndrome". Some believe PCV may be a variant of exudative age-related macular degeneration (AMD), while others believe it is an independent vascular entity.[2] It is commonly misdiagnosed as AMD or CSR due to its similar symptoms and clinical findings. These findings include SRF, subretinal hemorrhage (SRH), intraretinal fluid (IRF) and PEDs.[3,4]
PCV can be distinguished from AMD and CSR using indocyanine green angiography (ICG). Unfortunately, fundus fluorescein angiography (FFA), which is more readily available in clinical practices, often shows only non-specific PED and leakage that is present in a number of diseases.[2,5] The longer wavelength of light (~800 nm) used in ICG, compared to FFA (~490 nm), more effectively passes through the pigment of the retinal pigmented epithelium and therefore can better highlight the pathognomonic dilated choroidal vascular channels. These dilations appear as distinct, nodular "polypoidal lesions" and are best seen in the first six minutes of the ICG.[2] Additionally, fluorescein's lower affinity to plasma proteins as compared to indocyanine results in rapid choriocapillaris leakage and can cause confusion by showing polypoidal lesions as non-specific "hyperfluorescent spots" or revealing subretinal fibrinous material sometimes present in PCV as an expanding fluorescent leakage typically found in CSR and AMD.[2] Therefore, ICG is the preferred modality of imaging for PCV.
PCV is more common in Asian populations. The most common presenting symptom in PCV is decreased visual acuity (VA) due to SRF or SRH. Although the initial VA in PCV patients with SRF alone is usually better than patients with AMD, it is often more resistant to anti-VEGF therapy.[5] On fundus examination, the most suggestive findings of PCV are orange-red subretinal nodules, spontaneous SRH, and either serous or hemorrhagic PED.[2,5] The presence of one or more of these features suggest a diagnosis of PCV, and ICG should be performed.[2] However, these features are not always present in PCV or may not be readily appreciable and therefore ICG could be considered for all patients who fail to respond to anti-VEGF therapy.[4]
The polypoidal lesions are usually found in the perimacular area or close to serosanguinous PEDs and may form a "notch sign" between the polypoid lesion and the accompanying PED on OCT. Polypoidal lesions are not seen in AMD.[5]
Another typical OCT finding in PCV is a classic hyper-reflectivity in the choroidal layers known as the "double-layer" sign.[5] While PCV typically shows minimal or absent cystic change in the overlying retina, hard exudates and drusen may be present.[5] However unlike AMD, cases without exudative changes commonly occur in PCV[5].
PCV can be clinically classified as quiescent, exudative, or hemorrhagic. In quiescent disease, polyps exist without SRF, IRF, or hemorrhage.[2] Exudative disease requires SRF or IRF, but excludes hemorrhage.[2] Hemorrhagic disease includes all PCV with subretinal or sub-RPE hemorrhage.[2]
Studies have reported a better visual prognosis for PCV compared to exudative AMD, mainly due to spontaneous regression and the containment of PCV lesions under the RPE preventing intrusion and leakage in the subretinal space. Many patients, however, progress to significant and permanent vision loss.[5] A recent multicenter randomized controlled trial titled "EVEREST" (i.e. efficacy and safety of verteporfin photodynamic therapy in combination with ranibizumab or alone versus ranibizumab monotherapy in patients with symptomatic macular polypoidal choroidal vasculopathy) showed that treatment of PCV with a combination verteporfin photodynamic therapy (PDT) plus ranibizumab injections were well tolerated and highly effective in achieving complete regression of the polypoidal lesions in symptomatic PCV.[6] Specifically, combination PDT plus ranibizumab 0.5 mg monthly injections x3 showed complete polypoid regression in 77.8% of patients versus 71.4% for PDT alone and 28.6% for ranibizumab monotherapy.[6] Additionally, a long term visual outcome study of combination therapy showed that 88.2% of patients with PCV had either maintained or improved visual acuity 3 years after treatment.[7] In patients with incomplete regression of polypoidal lesions following initial combination treatment as assessed by ICG at 3 month follow-up, retreatment with PDT monotherapy or combination PDT/ranibizumab is recommended.[2,6] If either FFA or ICG still detects leakage and continued signs of disease activity, retreatment with only ranibizumab is appropriate.[2] Although ranibizumab monotherapy is effective in reducing subretinal fluid, it is much less effective in achieving polyp regression. Intravitreal monotherapy may be chosen over PDT/ranibizumab combination therapy in patients who have good visual acuity, or who have any contraindications to PDT (e.g. abnormal liver function tests).[2] There is insufficient evidence to support the use of corticosteroids in PCV.[2]
In summary, the clinical presentation of PCV is often similar to that of CSR or exudative AMD. The diagnosis of PCV can be challenging without ICG imaging. ICG should be considered in patients who have visible orange-red subretinal nodule(s), spontaneous massive subretinal hemorrhage, notched or hemorrhagic PED, or the lack of response to anti-VEGF therapy.2 In this case, it was the continued failure to respond to a variety of standard treatments for CSR that ultimately led to the ICG imaging and diagnosis of PCV. Additionally, because of our patient's good central VA, we pursued ranibizumab monotherapy and reserved possible PDT if the subretinal fluid does not improve or his VA decreases.
EPIDEMIOLOGY
|
SIGNS:
|
SYMPTOMS
|
TREATMENT
|
Suggested Citation format
Folchert M, Chin EK, Folk JC. Polypoidal Choroidal Vasculopathy (PCV). EyeRounds.org. May 4, 2014; available from https://eyerounds.org/cases/186-PCV.htm

Ophthalmic Atlas Images by EyeRounds.org, The University of Iowa are licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License.

Address
University of IowaLegal
Related Links
