
Chief Complaint: Bilateral corneal clouding noted after birth
A 2-day-old, 1920 gram infant born at 36 weeks gestation via caesarean-section due to suspected intrauterine growth restriction was evaluated for bilateral corneal opacity noted at the time of delivery (Figure 1). There were no known maternal infections during the pregnancy and no problems during the perinatal period. The mother had a herpetic oral ulcer, but no genital lesions. The infant did not show evidence of tearing, blepharospasm, photophobia, drainage, or ocular redness.
Fig. 1: Bilateral corneal opacities at 2 days of age. 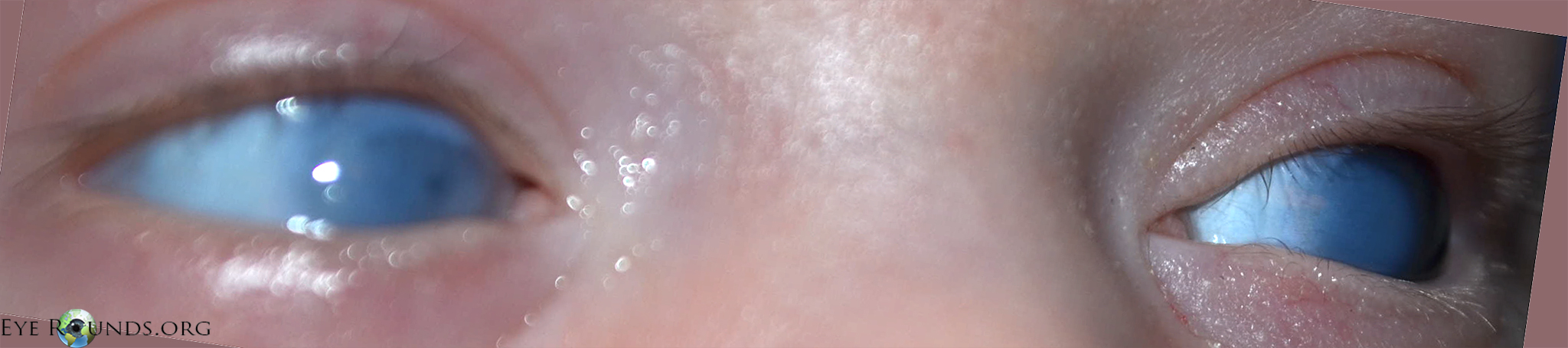
|
OD (right eye) |
OS (left eye) |
Intraocular pressure |
16 mmHg |
12 mmHg |
Corneal diameter |
8.5 mm |
9.0 mm |
Corneal pachymetry |
676 microns |
870 microns |
Retinoscopy |
Unable to perform |
+2.50 |
Fig. 2: Bilateral corneal opacities during examination under anesthesia. Note the more significant corneal opacification in the right eye compared to the left.
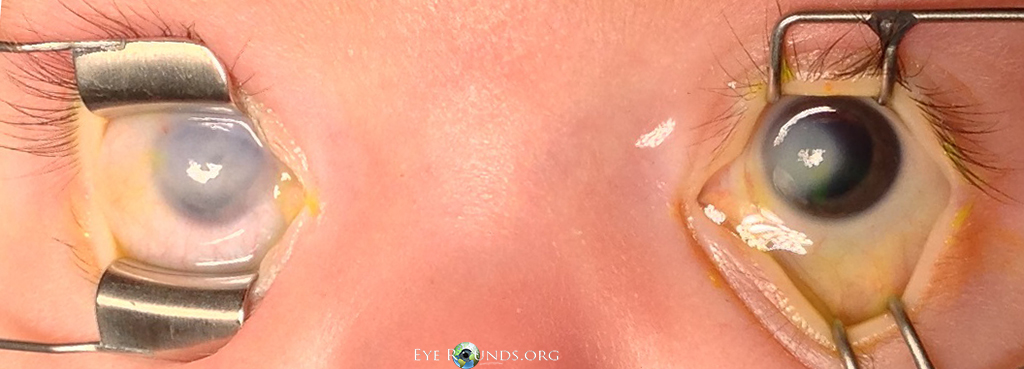
The decision was made to observe the left eye as the corneal opacification had improved at his 1 month visit and there was an excellent view to the posterior pole. However, to prevent amblyopia and provide visual rehabilitation a penetrating keratoplasty (PKP) of the right eye was recommended. The patient subsequently underwent synechiolysis of iridocorneal adhesions and PKP in the right eye. At his 4-month postoperative visit, the graft was clear and there was visual improvement OD. Genetic testing was also done for two autosomal-dominant genes for Peters anomaly, PITX2 and FOXC1 genes, but the results were negative.
Peters anomaly is a rare eye malformation resulting in congenital corneal opacity and is part of a spectrum of developmental anomalies of the cornea, iris and lens termed "anterior segment dysgenesis."
Peters anomaly was first described in 1906 by Albert Peters as a central corneal leukoma with variable synechiae between the iris and cornea and a defect in Descemet's membrane and the corneal endothelium. Since then, Peters anomaly has been subdivided into three types: 1) Peters type I characterized by central corneal opacity with iridocorneal adhesions (Figure 3), 2) Peters anomaly type II with central corneal opacity and cataracts or corneolenticular adhesions, and 3) Peters-plus syndrome with Peters anomaly and short stature, developmental delay, dysmorphic facial features including cleft lip/palate along with cardiac and genital abnormalities [1, 2]. Peters anomaly has a wide spectrum of severity and corneal opacification is bilateral in approximately 80% of cases [3].
Fig. 3: Small bilateral corneal opacities with associated iridocorneal adhesions in a patient with Peters anomaly.
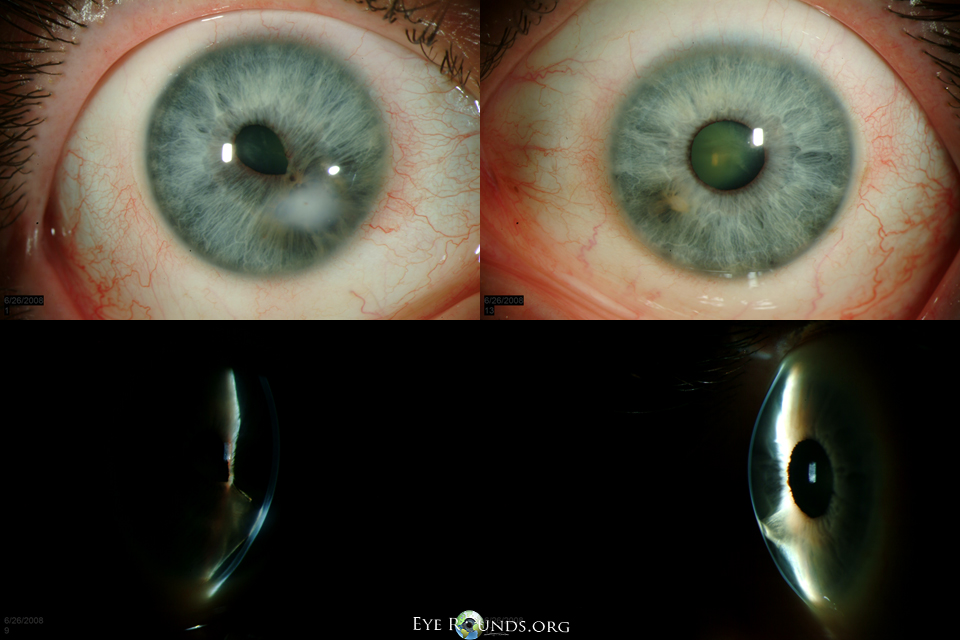
Patients with Peters anomaly are at increased risk of glaucoma, presumably due to abnormal development of the trabecular meshwork and Schlemm's canal and/or the presence of a shallow anterior chamber [1]. Approximately 50-70% of patients with Peters anomaly have associated glaucoma [3-5]. Glaucoma is more common in those with cataracts or corneolenticular adhesions. Elevated intraocular pressure (IOP) typically presents in infancy, but can also arise later in life.
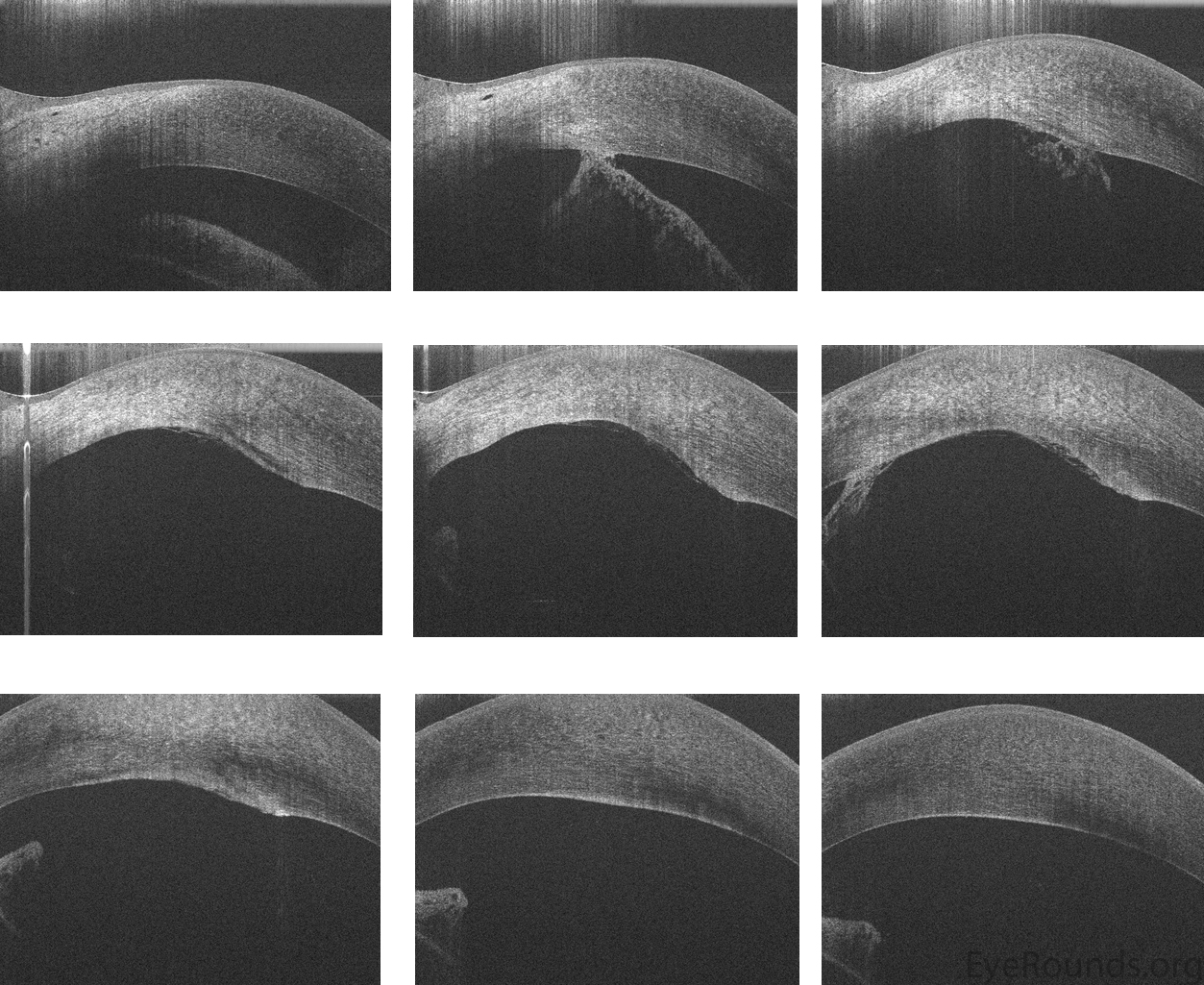
The diagnosis of Peters anomaly is made clinically with the finding of corneal opacification with corneal edema and underlying loss of Descemet's membrane and endothelium. Specular microscopy may be used to assess the presence and structure of these corneal layers. Ultrasound biomicroscopy (UBM) or anterior segment optical coherence tomography (AS OCT) can be performed to aid in the identification of iridocorneal or corneolenticular adhesions [2, 6] (Figure 4).
Fig. 4: Anterior segment optical coherence tomography (OCT) in a neonate with Peters anomaly. Note the iridocorneal adhesions and the region absent of normal posterior corneal structures.
Most cases of Peters anomaly are sporadic without an identified genetic cause. It can occur as an isolated finding or associated with systemic disease as in Peters-plus syndrome. Abnormal formation of the anterior segment causes an incomplete separation of the cornea from the iris or lens leading to corneal opacity. In the inherited forms, mutations causing abnormal migration of neural crest cells to the posterior cornea have been linked to Peters anomaly, including PAX6, FOXC1, PITX2 and CYP1B1. Mutations in these genes can also cause other clinical conditions such as aniridia, Axenfeld-Rieger syndrome, and primary congenital glaucoma (see differential diagnosis below). Mutations in PAX6, FOXC1, and PITX2 follow an autosomal dominant inheritance pattern while mutations in CYP1B1 follow an autosomal recessive pattern [1, 2].
Glaucoma associated with Peters anomaly is notoriously difficult to control and frequently requires surgical intervention. In a retrospective study, Yang et al. found that surgery was effective in controlling the IOP in only 32% of eyes at a median follow-up time of 11 years, even after multiple procedures [7]. There is little literature on the management of glaucoma associated with Peters anomaly and no head-to-head trials comparing the different surgical options. As a result, there is no established treatment algorithm for the condition. Based on the personal experiences of one of our senior glaucoma faculty members at the University of Iowa, patients are often initially managed medically. There are a variety of surgical options that can be attempted if medical management is insufficient. Trabeculotomy or goniotomy may be performed in eyes that have not undergone corneal surgery, though goniotomy can be difficult due to decreased visualization secondary to corneal opacification. In eyes that have already undergone corneal surgery, a glaucoma drainage device implant is often the initial surgery of choice if IOP control is unsatisfactory with medical management. Trabeculectomy is usually not preferred at very young ages and cyclodestructive procedures are reserved as a last option for glaucoma control when the visual prognosis is poor.
Children with corneal opacification or cataract involving the visual axis are at risk of developing amblyopia. Penetrating keratoplasty (PKP) or cataract extraction may be indicated to reduce this risk (Video). Visual prognosis is dependent on the disease severity. Although there is no formal classification for disease severity in Peters anomaly, Chang et al. defined patients with severe disease as being characterized by dense central opacities covering more than half of the cornea, corneolenticular adhesions, or associated ophthalmic anomalies like microphthalmia, aniridia, or cataracts. These patients had a visual acuity of finger-counting or worse and had poorer visual outcomes even with PKP. Patients with mild disease characterized by corneal opacities covering less than half of the cornea or iridocorneal adhesions and visual acuities of 20/100 or better had better visual outcomes with treatment [2]. Post-operatively there is also a role for amblyopia therapy to improve visual outcomes [8].
Video: Penetrating keratoplasty (PKP) in a 2-year-old boy with bilateral corneal opacification and lenticular adhesion due to type II Peter's anomaly. Unfortunately, there was near-immediate vitreous loss in this case requiring anterior vitrectomy. If video fails to load, view on YouTube
However, success rates for pediatric PKP is much lower than for adults. Several studies have shown a wide range of graft failure rates at 1 year ranging from 22%-67% [9-12]. Some of this variability may stem from a higher success rate of PKP in patients with Peters type I (87.5%) versus Peters type II (14.2%) [1]. Yang et al. showed that less than one-third of Peters anomaly patients undergoing PKP achieved visual acuity of 20/400 or better [13].
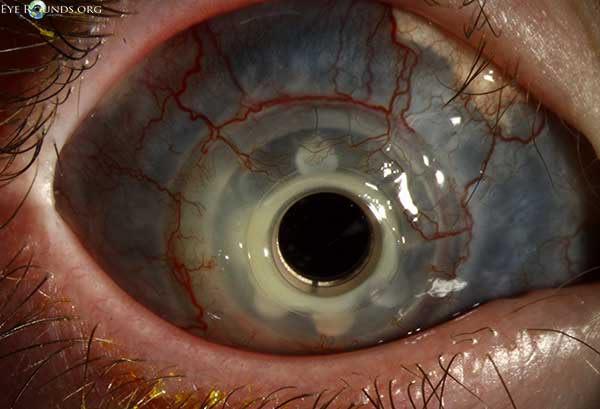
Due to the high prevalence of PKP graft failure, some have advocated for the use of the Boston Type I Keratoprosthesis (KPro-1) in these patients (Figure 5). Aquavella et al. tracked outcomes in 13 patients with Peters anomaly undergoing receiving KPro and found that the prosthesis was well-tolerated and retained without dislocation or extrusion at a mean follow-up period of 9.7 months [14]. This study highlights that KPro may be a viable alternative to PKP.
Therefore overall prognosis and the decision to intervene for Peters anomaly depends heavily on disease severity. Patients with severe disease had poorer visual outcomes compared to those with mild disease regardless of treatment [2]. This may be due to already poor visual acuity prior to treatment that is unlikely to change with surgery. However it is unclear which treatment modality is superior. Further studies are needed to compare the long-term outcomes between PKP and KPro implants in children.
Fig. 5: Post-operative appearance of a Boston Type I Keratoprosthesis (KPro-1) after multiple failed penetrating keratoplasties in a patient with Peters anomaly.
A useful mnemonic that is commonly used for the differential of a cloudy cornea at birth is STUMPED:
Sclerocornea
Tears in Descemet's membrane (forceps injury or Haab's striae)
Ulcer
Metabolic
Peters anomaly
Edema (congenital glaucoma or CHED)
Dermoid
Chang E, Vislisel J, Larson, SA. Peters Anomaly. EyeRounds.org. June 30, 2014; available from https://eyerounds.org/cases/187-Peters-Anomaly.htm

Ophthalmic Atlas Images by EyeRounds.org, The University of Iowa are licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License.

Address
University of IowaLegal
Related Links
