
A 79-year-old woman was referred for the evaluation of a corneal ulcer in the right eye (OD). She began experiencing foreign body sensation in the right eye about 2 months prior to presentation after getting poked in the eye while gardening. Since that time, she had experienced pain, tearing, photophobia, and decreased vision in the eye. The patient was treated with prednisolone acetate 1% drops every 2 hours in the right eye locally, but this was discontinued as corneal thinning was noted. She was a poor historian but apparently carried a diagnosis of "rheumatoid arthritis" for which she chronically took 5 mg of oral (PO) prednisone daily. This was increased to 60mg PO daily by her local ophthalmologist about 2 weeks prior to her referral. She had never seen a rheumatologist.
Pseudophakia, both eyes (OU). History of cataract extraction in both eyes 3 years ago.
No family history of rheumatologic or significant ocular disease.
Hypertension, possible "rheumatoid arthritis," dementia
Moxifloxacin (Vigamox) twice daily, OD
Oral Prednisone 60 mg daily for the past 12 days, furosemide, metoprolol
The patient is retired. She has occasional alcohol use. There is no history of tobacco or illicit drug use.
Complete review of systems is negative except as in HPI.
OD: 20/200
OS: 20/40+2
Pupils: Equal and round with brisk light response and no relative afferent pupillary defect OU.
Confrontation visual fields: Full to counting fingers OU
Extraocular movements: Full OU
Intraocular pressure (Tonopen):
OD: 17 mmHg
OS: 17 mmHg
OD:
- Eyelid: Meibomian gland dysfunction
- Conjunctiva: 1+ injection superiorly
- Cornea: Crescent-shaped ulceration of peripheral cornea between 11:00 and 3:00 with associated epithelial defect and 80% stromal thinning.
- Anterior chamber: 3+ cell, trace flare
- Iris: Normal architecture
- Lens: Posterior chamber intraocular lens
OS:
- Eyelid: Meibomian gland dysfunction
- Conjunctiva: No injection
- Cornea: Clear and compact
- Anterior chamber: Deep and quiet
- Iris: Normal architecture
- Lens: Posterior chamber intraocular lens
Fundus exam: Normal OU
A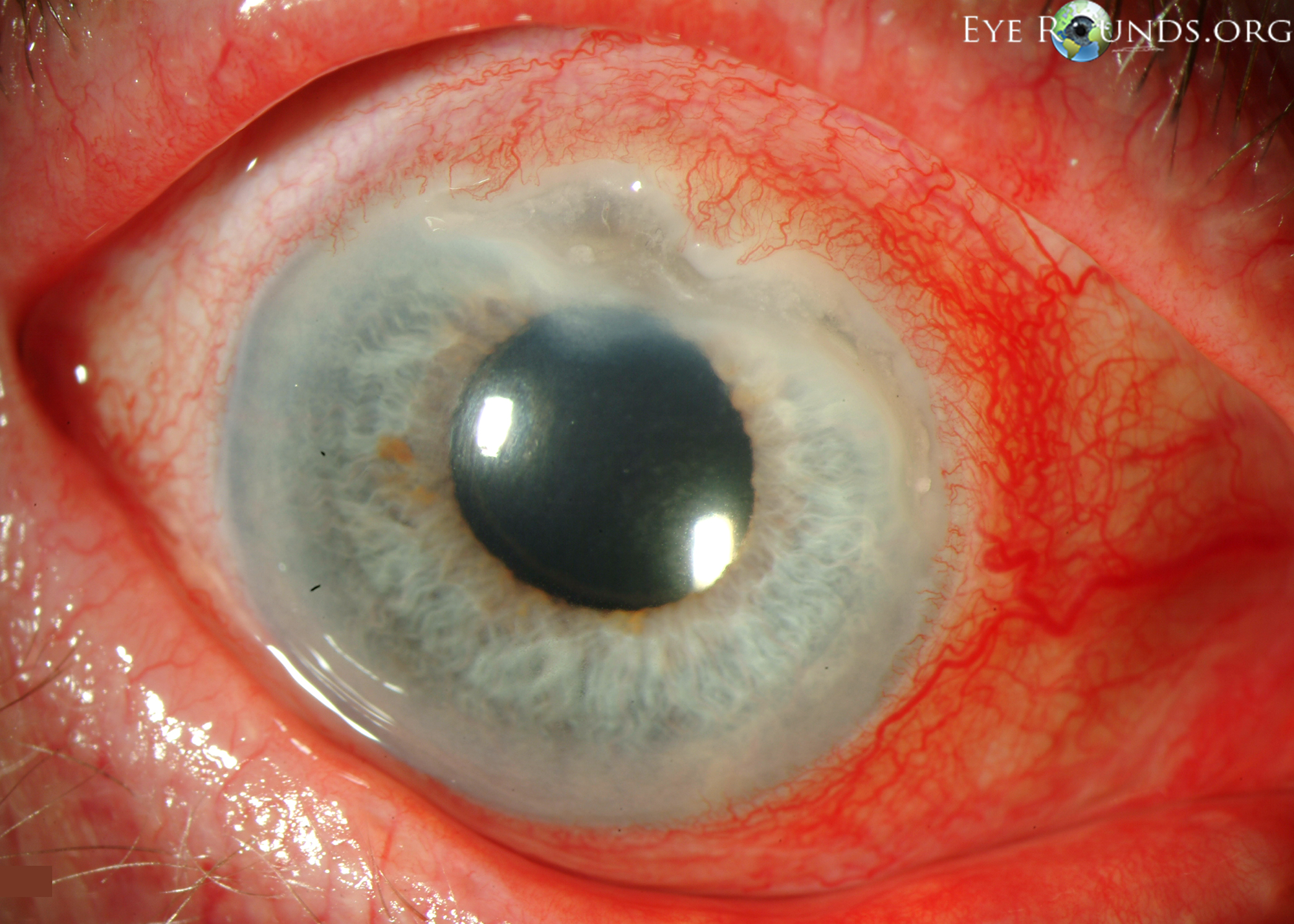 |
B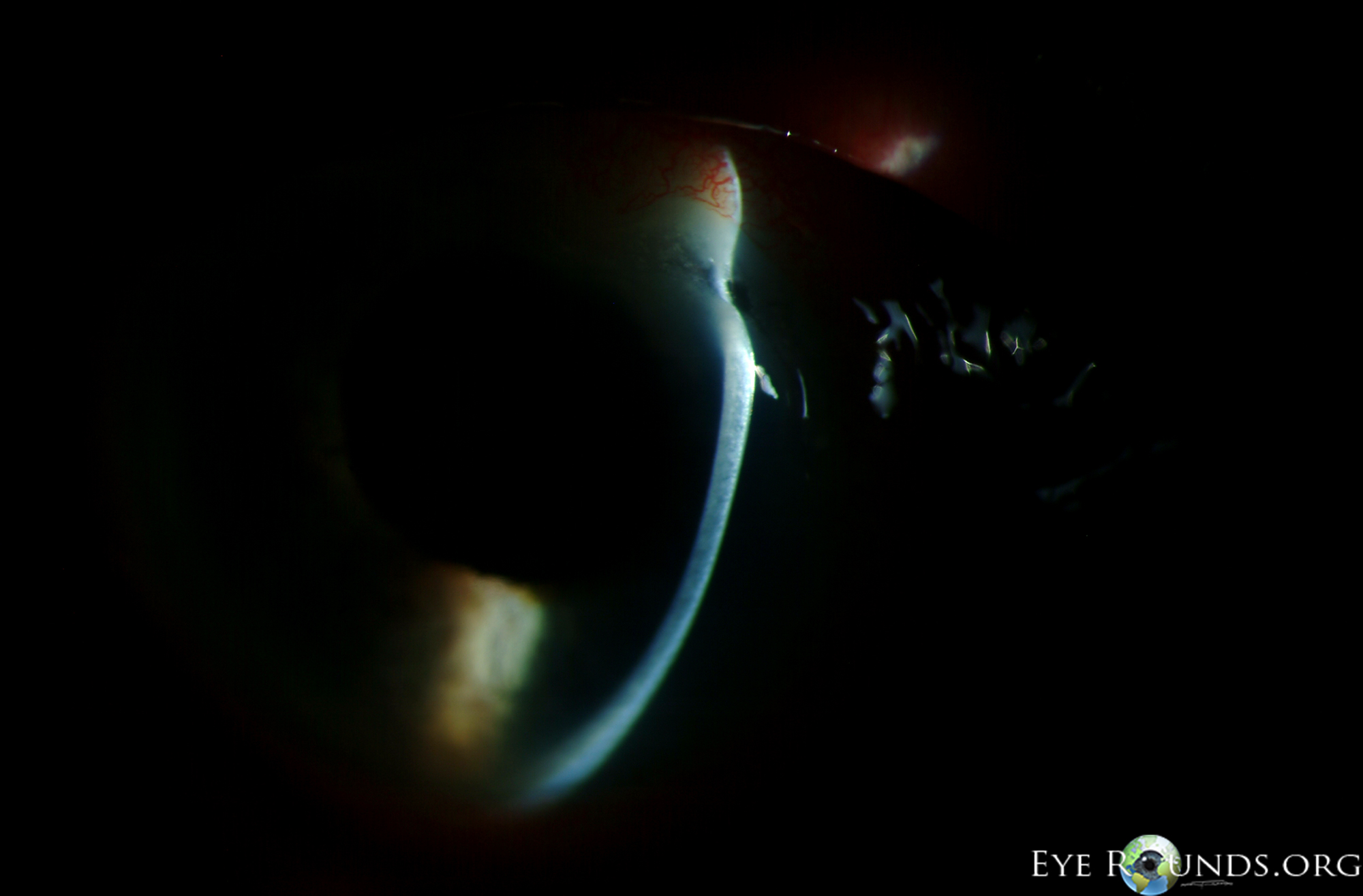 |
C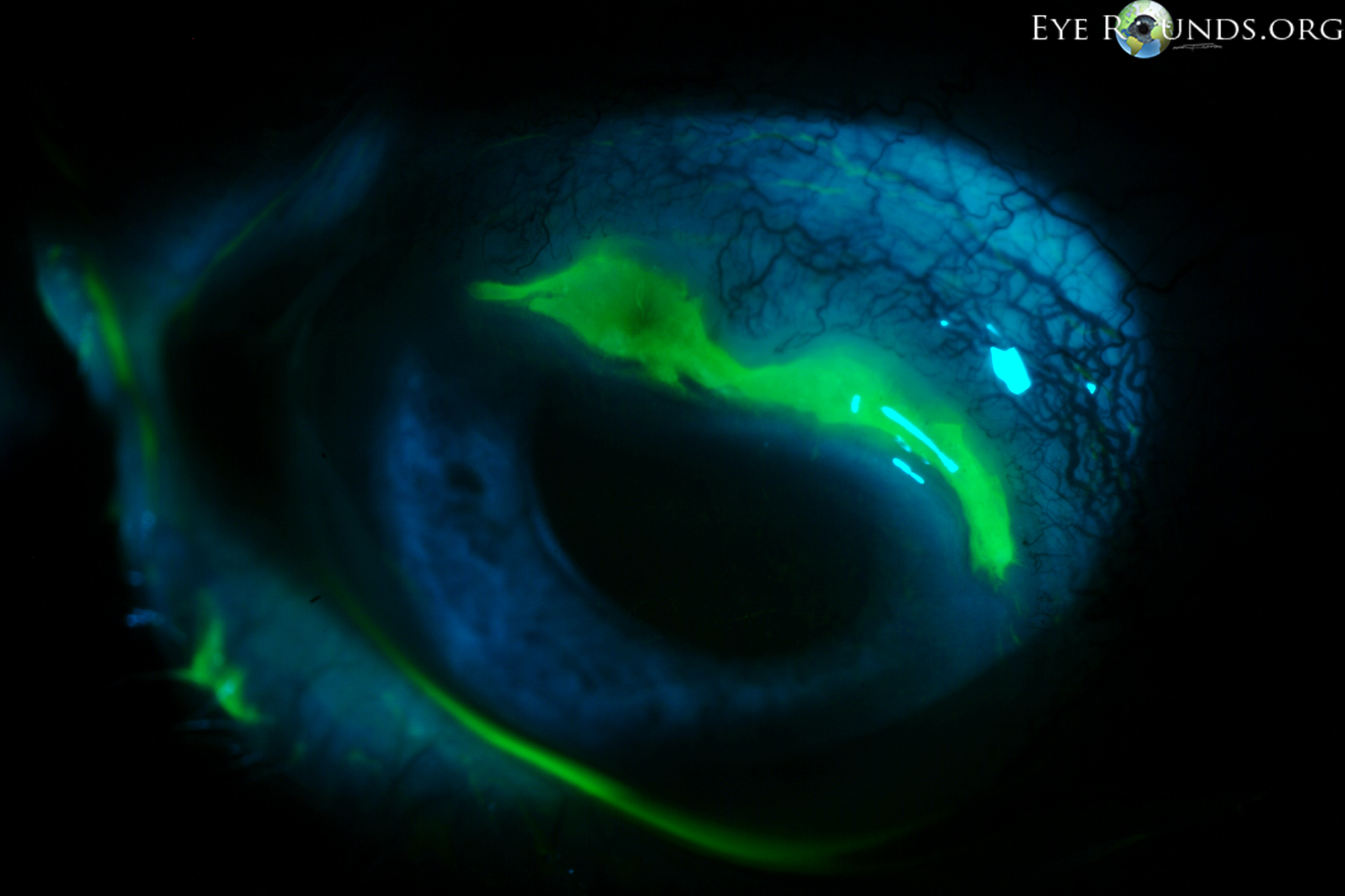 |
Figure 1: Slit lamp photographs (A, B) showing a superior, peripheral, crescent-shaped corneal ulcer with 80% corneal thinning OD. There is adjacent conjunctival injection. The fluorescein stain (C) more clearly reveals the area of epithelial defect.
The patient presented to the University of Iowa Hospitals and Clinics (UIHC) with a peripheral corneal ulceration in the right eye in the setting of arthritis and an unclear history of rheumatologic disease. The presentation was most consistent with peripheral ulcerative keratitis (PUK) with corneal melt resulting in prominent corneal thinning. The anterior chamber cell and flare was believed to be reactive inflammation secondary to the corneal process.
We contacted her primary care physician who stated she had "polyarteritis" diagnosed 5 years ago that was "rheumatoid factor negative." In addition to the chronic daily oral prednisone, she was supposed to be taking methotrexate 20 mg every 4 weeks, but regularly missed her appointments for the injections. Her last injection was 8 weeks ago. She had never seen a rheumatologist.
Blood work for associated systemic conditions was drawn including complete blood count (CBC), blood urea nitrogen (BUN), creatinine, erythrocyte sedimentation rate (ESR), rheumatoid factor (RF), antinuclear antibodies (ANA), antineutrophil cytoplasmic antibodies (ANCA), anti-Ro and anti-La antibodies, and angiotensin-converting enzyme (ACE). A formal consultation was made with rheumatology. We continued moxifloxacin (Vigamox) BID OD for infection prophylaxis and initiated therapy with cyclosporine 0.05% (Restasis) BID OD and doxycycline 100 mg PO daily for their anti-inflammatory effects. Prednisone was continued at 60 mg PO daily. A bandage contact lens was placed on the eye for comfort.
The laboratory workup was significant for elevated c-ANCA which led to a diagnosis of Wegener's granulomatosis by the patient's local rheumatologist. She was started back on weekly doses of methotrexate and her prednisone was tapered with resolution of her keratitis.
Peripheral ulcerative keratitis (PUK) is an inflammatory corneal condition estimated to affect 3 persons per million per year (1). It predominantly affects older individuals and does not have a significant gender predilection (2,3,4). The condition may be triggered by surgical or non-surgical trauma, or may arise spontaneously (3). Patients often present with ocular redness, pain, tearing, photophobia, and decreased vision. The typical exam finding is a crescent-shaped corneal ulceration (stromal thinning with overlying epithelial defect) located in the peripheral cornea within 2 mm of the limbus (5). The predilection for the periphery is believed secondary to the close proximity to the conjunctival vessels which supply the inflammatory mediators that cause the disease. There may be surrounding infiltrate and localized conjunctival injection adjacent to the ulcer. Cases can be bilateral, but are more commonly unilateral (2,3). Scleritis occurs simultaneously with PUK in 36-66% of cases. Necrotizing scleritis is most common and one should be vigilant for this in any patient with both scleritis and PUK (2,3). Anterior uveitis also frequently accompanies PUK and is present in 9-67% of patients (2,3).
PUK is an immunologic condition mediated by both abnormal T-cell and antibody-mediated pathways (6). It is hypothesized that an abnormal T-cell pathway may produce antibodies that result in immune-complex deposition in the cornea and later activate the complement system and recruit harmful inflammatory cells to the area. Neutrophils and macrophages then secrete local collagenases and other proteases which cause destruction of the corneal stroma.
At least half of PUK cases are associated with systemic immune-mediated or rheumatic diseases (1,2). In a quarter of cases, PUK may be the initial manifestation of disease (2). The most common systemic association is rheumatoid arthritis, which is present in 34-66% of patients (2,3,4). In these patients, PUK occurs late in the disease course, with a mean onset 19.6 years after diagnosis (1). PUK develops earlier with Wegener's granulomatosis, often presenting within 6 months of the diagnosis, or precluding the diagnosis as it did in our patient (1). Other associated systemic conditions include Churg-Strauss syndrome, microscopic polyangiitis, polyarteritis nodosa, relapsing polychondritis, and systemic lupus erythematosus. Understanding common manifestations of these diseases can be helpful. For example, a history of epistaxis or sinusitis may be evidence of Wegeners granulomatosis, while a history of asthma or atopy may be present in cases of Churg-Strauss.
PUK can also be associated with local or systemic infectious processes including those caused by bacteria (Staphylococcus and Streptococcus), spirochetes (Treponema pallidum), mycobacteria (tuberculosis), viruses (hepatitis C, herpes simplex virus, varicella zoster virus), Acanthamoeba, and fungi (Table 1)(6).
Table 1: Local and systemic diseases associated with PUK (2,6) |
||
Autoimmune diseasesVasculitides
Other autoimmune
|
InfectionsBacterial
Viral
Protozoal
Fungal |
OtherOcular
Dermatologic
Other systemic
|
Because of the high association with potentially lethal systemic diseases, all patients with PUK of unknown cause should undergo systemic workup including thorough review of systems in addition to laboratory and radiologic studies (Table 2) (5).
| Table 2: Laboratory and radiologic evaluation for patients with PUK (5) | |
Standard evaluationCorneal cultures for bacteria, viruses, and fungi Complete blood count (CBC) Complete metabolic panel (CMP) Urinalysis with microscopic analysis (UA with micro) Antineutrophil cytoplasmic antibody (ANCA) Antinuclear antibody (ANA) Rheumatoid factor (RF) Anticyclic citrullinated peptide (anti-CCP) antibody Chest radiograph (CXR) Rapid plasma reagin (RPR) Fluorescent treponemal antibody absorption (FTA/ABS) Lyme antibody |
Additional tests as guided by history and physical examinationTuberculin skin test Sacroiliac joint radiographs Sinus imaging Viral hepatitis panel |
Wegener's is a small vessel vasculitis that affects mostly the lungs and kidneys. Pathology demonstrates giant cells with necrotizing granulomas. Classic signs and symptoms include pulmonary infiltrates (71%), sinusitis (67%), epistaxis, rhinorrhea, and hemoptysis. Vague symptoms are common such as joint pain, fatigue, and weight loss (18). The American College of Rheumatology (ACR) diagnostic criteria have shown to be 88% sensitive and 92% specific in the diagnosis of Wegener's granulomatosis (Table 3) (19).
Table 3: ACR clinical criteria for the diagnosis of Wegener's granulomatosis (requires 2 of 4) |
|
Ocular involvement with Wegener's is quite common and is a presenting symptom in up to 16% of cases (Figure 2, Table 4) (18,19). Children more frequently present with ocular involvement than adults (20). Ocular involvement has been cited to be as high as 58% at some point in the clinical course (21).
A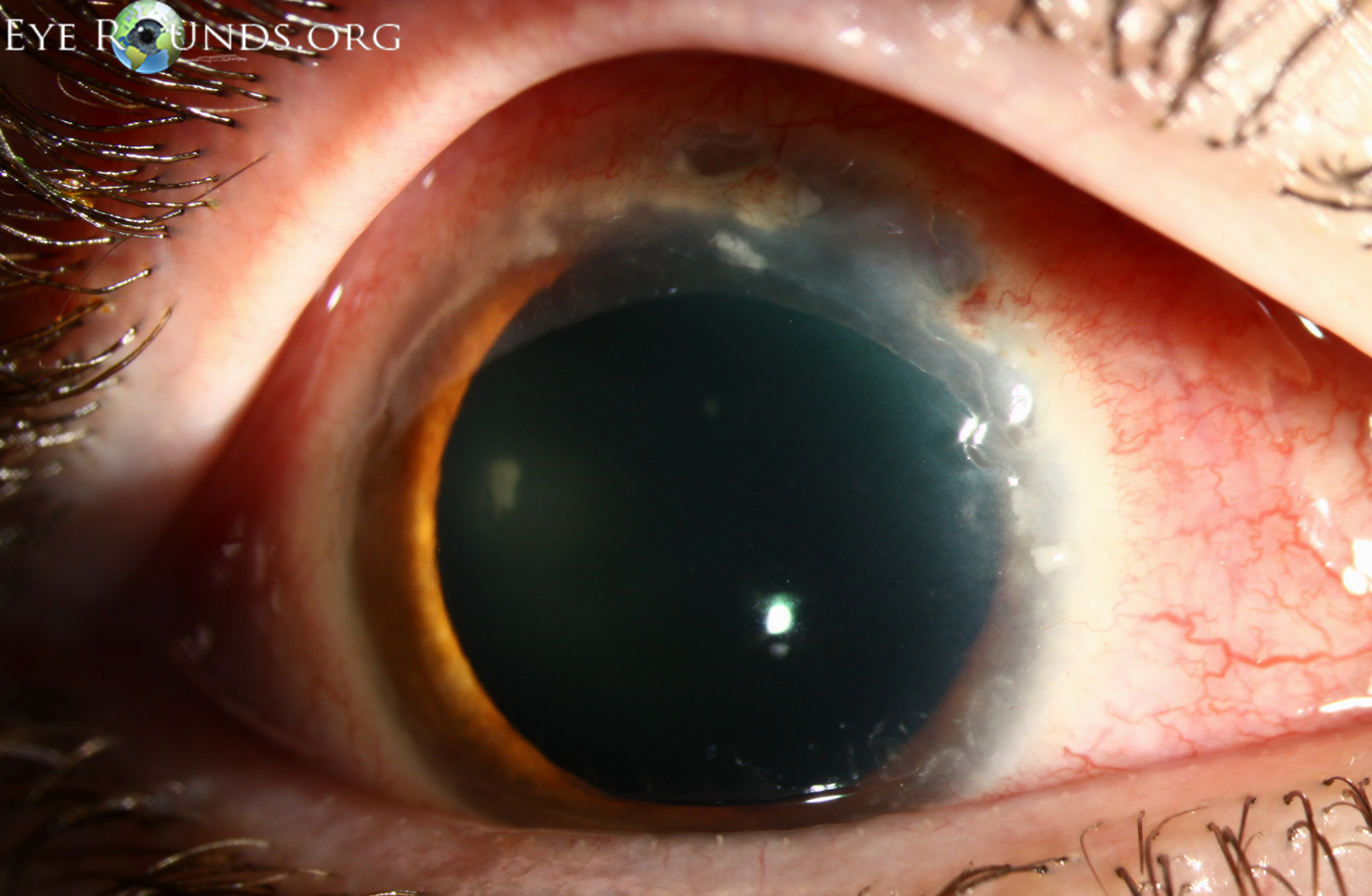 |
B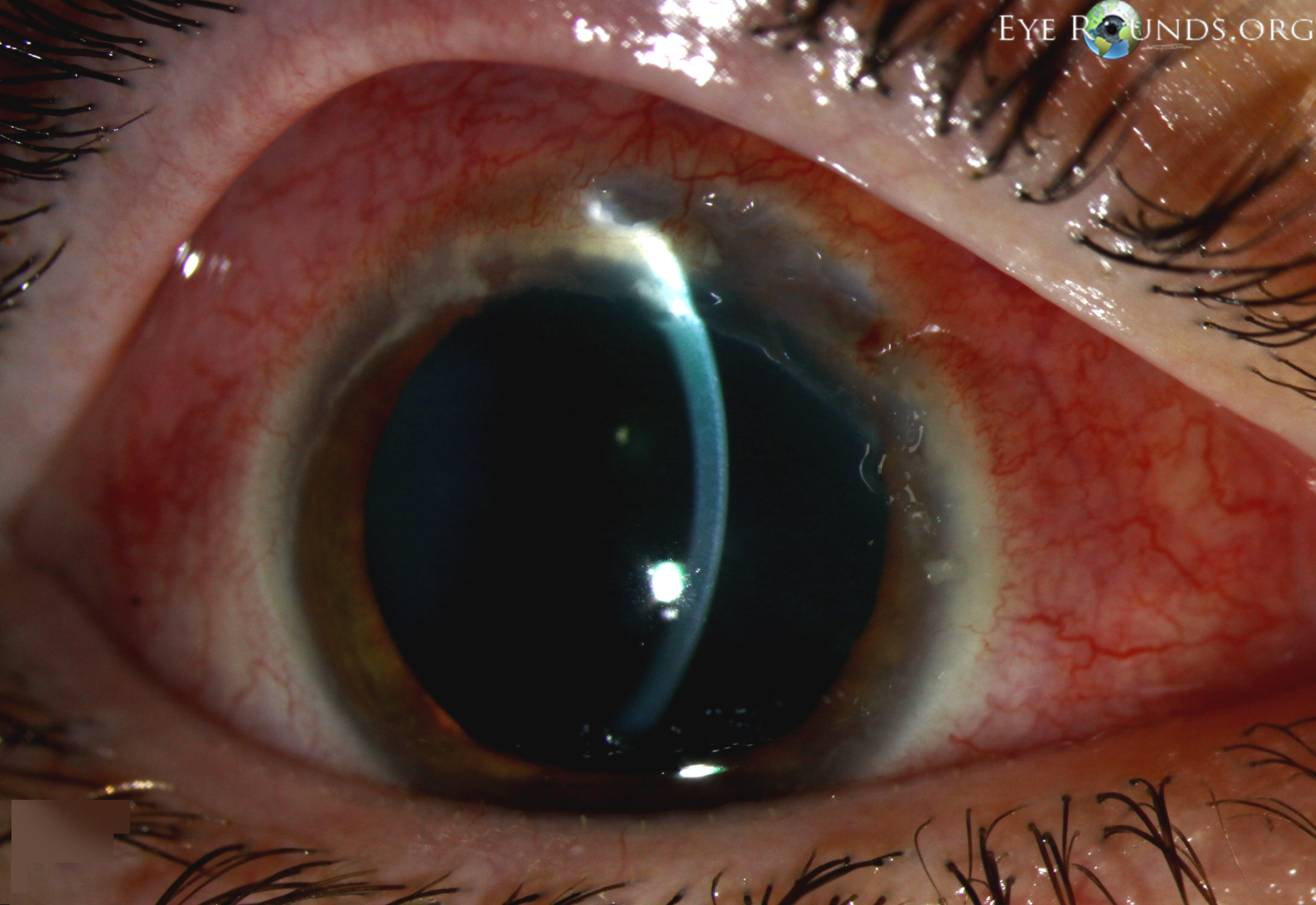 |
C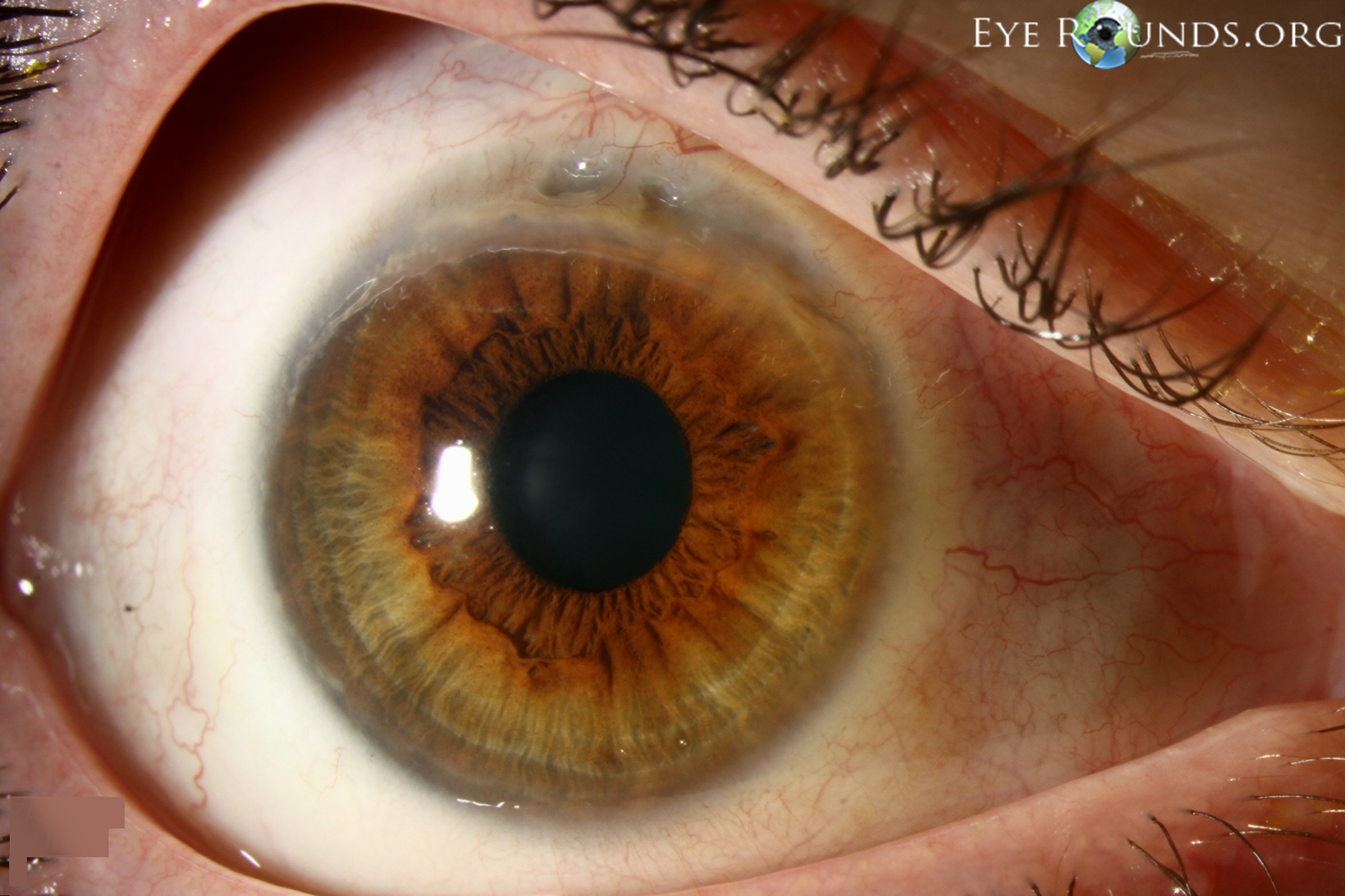 |
Figure 2: A different patient who was diagnosed with Wegener's granulomatosis after presenting with PUK. Review of systems revealed a 3-month history of joint pains, generalized fatigue, bloody bowel movements, and sinus discharge. Lab workup revealed mild normocyctic normochromic anemia with elevated ESR, CRP, and c-ANCA. The urinalysis showed proteinuria and microhematuria. (A, B) Superior epithelial defect with 80% stromal thinning and adjacent sterile corneal infiltrates. (C) Clinical appearance 1 week after initiating treatment. The eye appears less inflamed with resolution of corneal infiltrates.
Table 4: Ocular manifestations of Wegener's granulomatosis (23) |
|
Anti-neutrophil cytoplasmic antibody (ANCA) autoantibodies are very helpful in making the diagnosis of Wegener's. The c-ANCA test is quite specific for the disease process (93% sensitivity, 97% specificity) (22). The p-ANCA autoantibody can also be seen with this disease process, but it is also seen in other vasculitides such as microscopic polyangiitis. Some authors suggest that these autoantibodies can be used for monitoring disease activity as these tests will change with degree of activity of the disease process. Other common laboratory findings in Wegener's include an elevated ESR, positive rheumatoid factor, and mild normocytic anemia. Antinuclear antibodies (ANA) are normally negative (18).
PUK can lead to serious ocular complications (2,3,4,7). The corneal ulcer can progress rapidly, sometimes leading to corneal melt syndrome (keratolysis) and ocular perforation within days. This dire complication occurs in 14-35% of cases (4,7). Around half of patients may require urgent corneal surgery (2,7). Many of the eyes can be saved, but one report found that nearly 10% of eyes are lost despite treatment (7). Permanent reduction in vision results from corneal scarring (2,3,4,7). Between 3-35% have a visual acuity worse than 20/200 at most recent follow-up (4,7).
In addition to the ocular complications, PUK may be a sign of more widespread vasculitis which can cause significant problems with other organ systems and overall mortality of up to nearly 30% (8,9).
It is important to recognize and treat patients with PUK due to the high potential for ocular complications and the high mortality rate in the presence of associated systemic disease (8). Recently, there has been a changing trend in the treatment of autoimmune and vasculitic diseases in which second-line systemic immunosuppressive medications and monoclonal antibody treatments are implemented earlier in the treatment of disease (4,10).
The treatment of PUK depends on the severity of ocular and associated systemic disease. The primary treatment for acute disease control is systemic corticosteroids, often started at 0.5-1 mg/kg/d with a maximum dose of 60-80 mg daily (5,7,11). Patients with imminent vision loss may be started initially on pulsed methylprednisolone 1 g/d for 3 days and then transitioned to oral steroids. The steroids are tapered in accordance with the activity of the disease. Usually, prednisone is given at a high dose for 2-4 weeks then tapered over the course of several months (18).
An additional immunomodulatory agent is often started concurrently with the corticosteroids in the treatment of PUK. The choice of immunosuppressive medication depends on the underlying systemic disease process and its severity. The first-line for rheumatoid arthritis-associated PUK is often methotrexate (5-25 mg once weekly) (9). If this proves ineffective, other immunomodulatory agents such as cyclophosphamide, azathioprine, cyclosporine, or anti-tumor necrosis factor (TNF) agents may be utilized. Wegener's granulomatosis may require more powerful immunosuppressive medications for control. Cyclophosphamide (initial dosage of 2 mg/kg) is often first-line for Wegener's granulomatosis-associated PUK (9). Azathioprine is a less-effective alternative for those who do not tolerate cyclophosphamide. In addition to controlling ocular and systemic disease, cytotoxic agents have also been shown to prolong corneal graft survival (12) and reduce overall mortality (8). Unfortunately, relapse is quite common in these disease processes.
Patients on immunosuppressive medications require frequent exams and blood work to monitor for medication-induced complications. Patients on both corticosteroids and cytotoxic agents should be on prophylaxis against Pneumocystis jiroveci pneumonia (PCP). A common regiment for this is a trimethoprim-sulfamethoxazole single-strength tablet daily (5). Calcium and vitamin D supplementation and bone density monitoring should be considered for patients on chronic corticosteroids. Rheumatology consultation is usually advised.
Local therapy should include aggressive lubrication and eyelid hygiene, possibly incorporating the use of punctal plugs (9). Lubrication is useful as many cases have a component of keratoconjunctivitis sicca and it also may dilute inflammatory mediators in the tear film. Oral doxycycline can assist in ocular surface lubrication through its effects on the Meibomian gland secretions and also has anti-inflammatory effects. Topical cyclosporine 0.05% (Restasis) has been reported to be successful for non-healing ulcers, likely due to its anti-inflammatory action (13). Topical corticosteroids suppress corneal inflammation, but they are usually best avoided in cases of PUK as they can contribute to corneal melt by suppressing collagen production. Topical antibiotics should be utilized for infectious prophylaxis as long as an epithelial defect exists. The long-term use of moxifloxacin (Vigamox) drops may seem safe due to the lack of preservative, but prolonged exposure to the fluoroquinolone class of antibiotics may actually enhance melting by increasing matrix metalloproteinases in the cornea. Bacteriostatic antibiotics such as trimethoprim/polymyxin B (Polytrim) may be preferred for long-term use.
In cases of frank or impending corneal perforation, surgical intervention is warranted as a temporizing measure to save the eye. Surgery alone, however, does not resolve the underlying inflammatory process and does nothing to prevent recurrences of ocular involvement (9). There are multiple surgical options to maintain structural integrity of the globe. Cyanoacrylate adhesive helps arrest corneal melting by limiting the access of inflammatory cells to the corneal stroma (14). Penetrating keratoplasty or lamellar grafts can be utilized for structural support or visual rehabilitation (Figure 3, Video 1). Penetrating keratoplasty in patients with PUK has been shown to have a high failure rate, most commonly due to graft melt from PUK recurrence (15,16). Reports have shown 20-40% graft survival at 6 months and many patients therefore require multiple grafts (12,17). Systemic cytotoxic agents reduce the rate of graft failure by treating the underlying inflammatory condition and possibly reducing the rate of endothelial graft rejection (12). Bandage contact lenses may be utilized in less severe cases or in combination with the above procedures. Some have advocated for surgical conjunctival resection adjacent to the ulcer to decrease the access of inflammatory cells and factors to the cornea, but this remains controversial as it has not been definitively demonstrated to alter the course of the disease (2,9). Tarsorrhaphy may be helpful if exposure is playing a role.
A 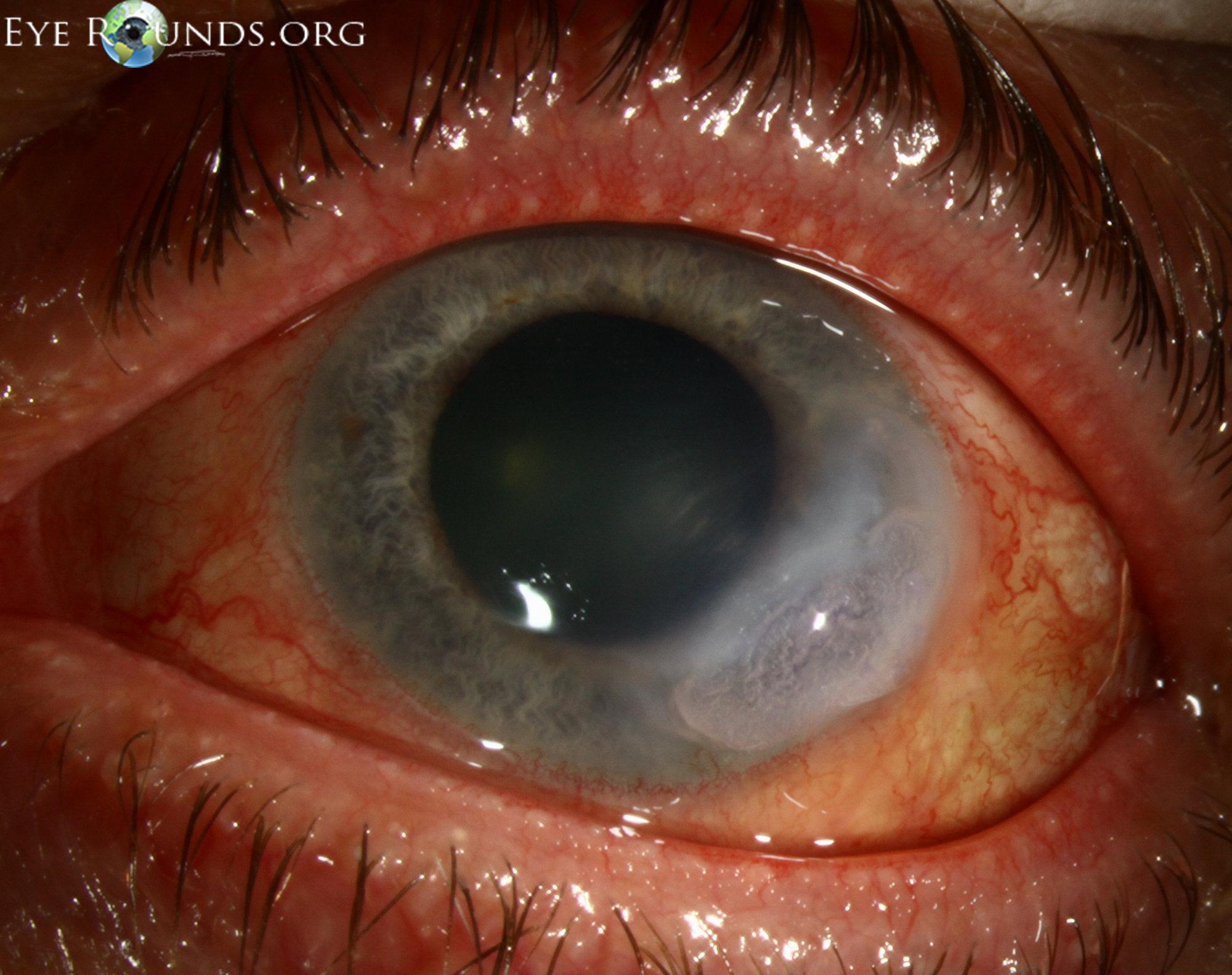 |
B 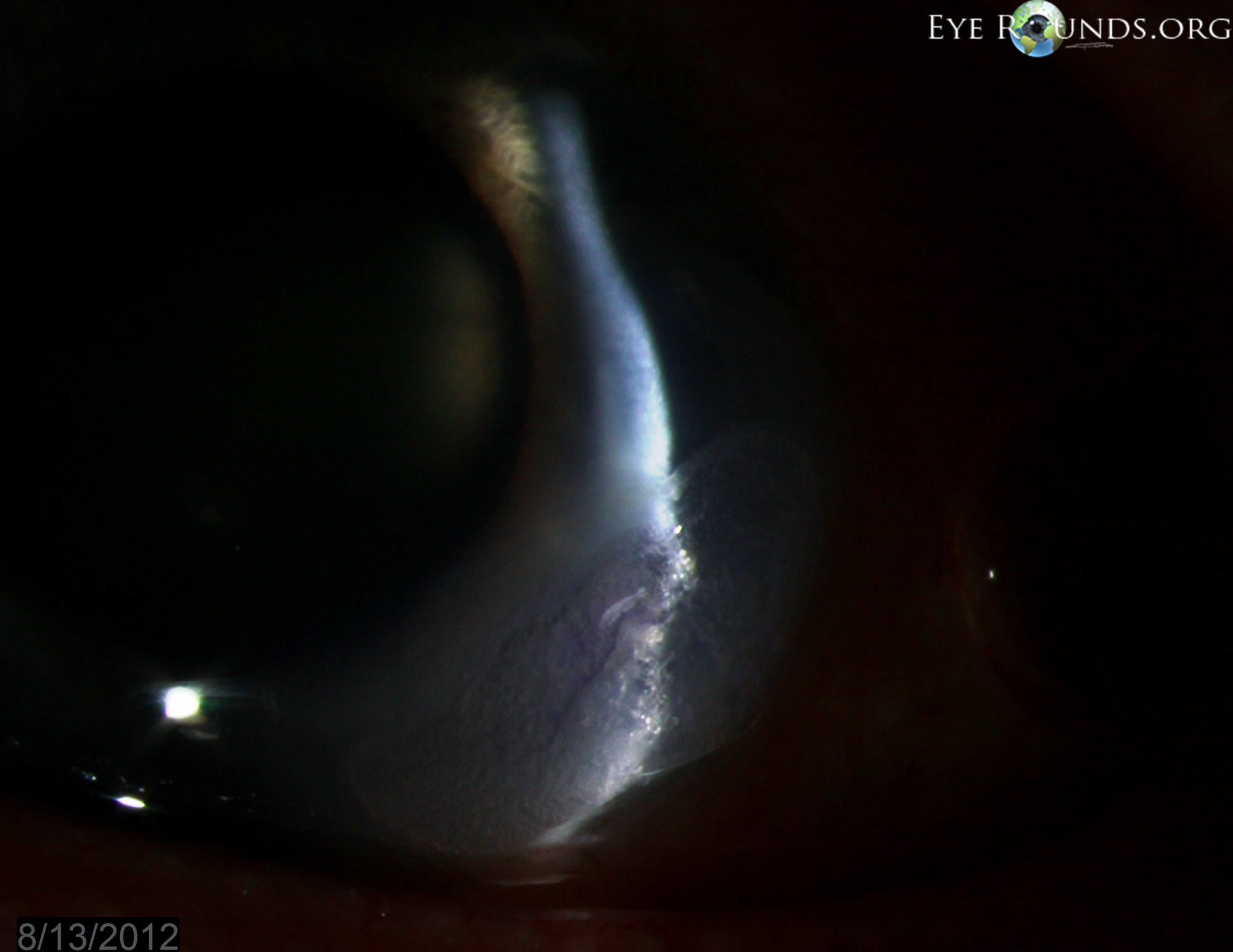 |
C 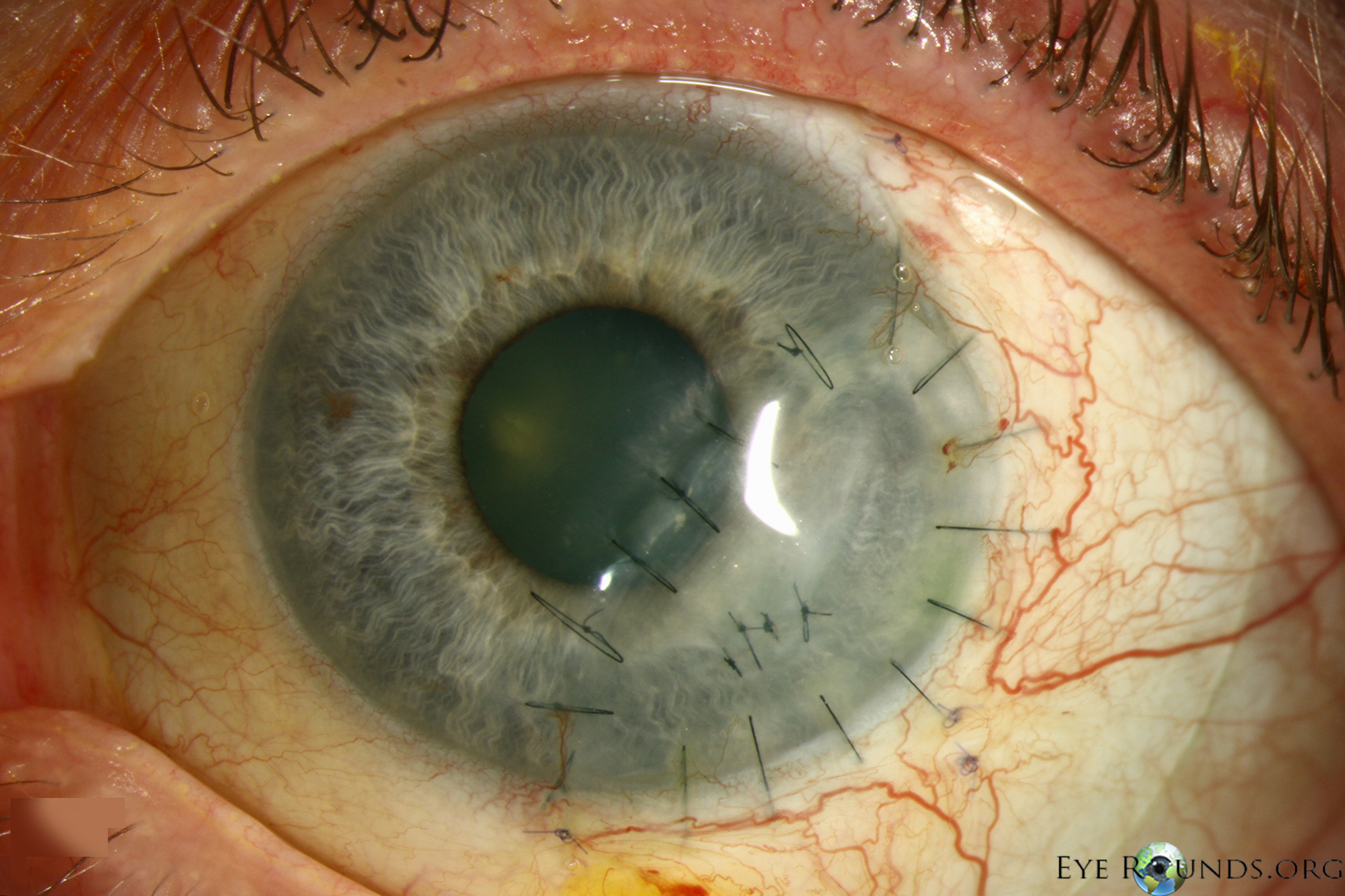 |
Figure 3: This patient with PUK in the setting of lupus and ocular rosacea presented with a leaking descemetocele (A, B) and underwent a lamellar keratoplasty to restore structural integrity to the eye (C). A video of the surgical procedure can be viewed in video 1.
Video 1: Tectonic anterior lamellar keratoplasty for peripheral corneal perforation in the patient pictured in Figure 2. If video fails to load, use this link.
Peripheral ulcerative keratitis (PUK) associated with Wegener's granulomatosis
EPIDEMIOLOGY
|
SIGNS
|
SYMPTOMS
|
TREATMENT
|
Vislisel JM, Lenci L, Goins KM. Peripheral Ulcerative Keratitis (PUK). EyeRounds.org. August 25, 2014; Available from: https://eyerounds.org/cases/194-PUK.htm

Ophthalmic Atlas Images by EyeRounds.org, The University of Iowa are licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License.

Address
University of IowaLegal
Related Links
