
The patient is a three-year-old male who was referred to the cornea clinic after failing a KidSight screening and was found to have bilateral corneal opacities when evaluated by his optometrist. Otherwise the child is in good health without any known medical problems.
Past Ocular History: He has a history of nasolacrimal duct obstruction with spontaneous resolution. He has no history of eye crossing or drifting.
Past Medical History: He has an unremarkable birth history and is otherwise healthy.
Medications: None
Family History: He has a paternal grandmother with glaucoma. No other known family history of eye disease or corneal dystrophies.
|
Right |
Left |
|---|---|---|
Visual Acuity (LEA symbols, without correction) |
20/25 |
20/20 |
Intraocular Pressure (IOP) |
15 mmHg |
15 mmHg |
Pupils |
Round and regular; reactive with no relative afferent pupillary defect (RAPD) |
Round and regular; reactive with no RAPD |
Confrontational Visual Fields |
Full to toys |
Full to toys |
Extraocular Motility |
Full |
Full |
Lids/Lashes |
Normal |
Normal |
|---|---|---|
Conjunctiva/Sclera |
Clear and quiet |
Clear and quiet |
Cornea |
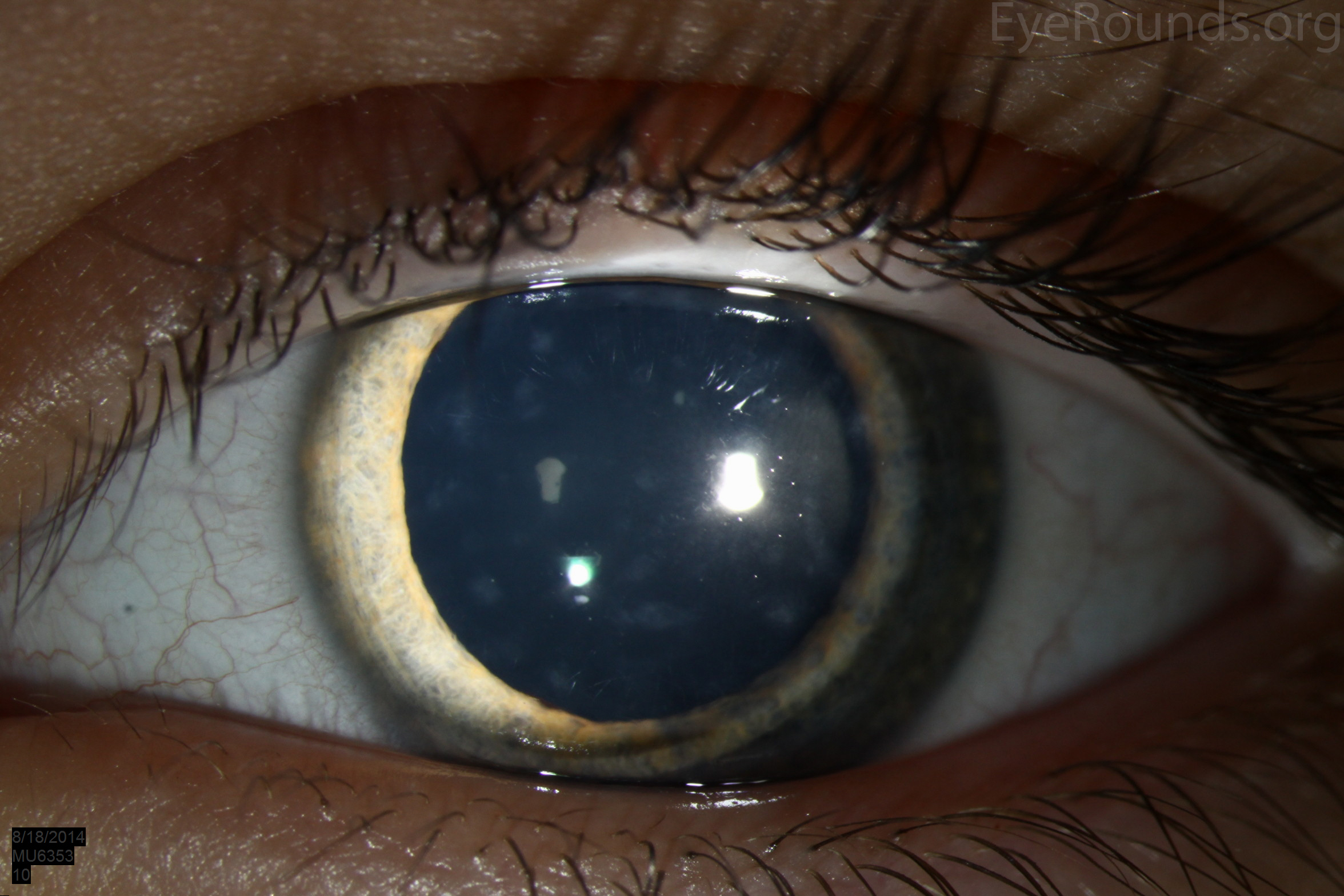
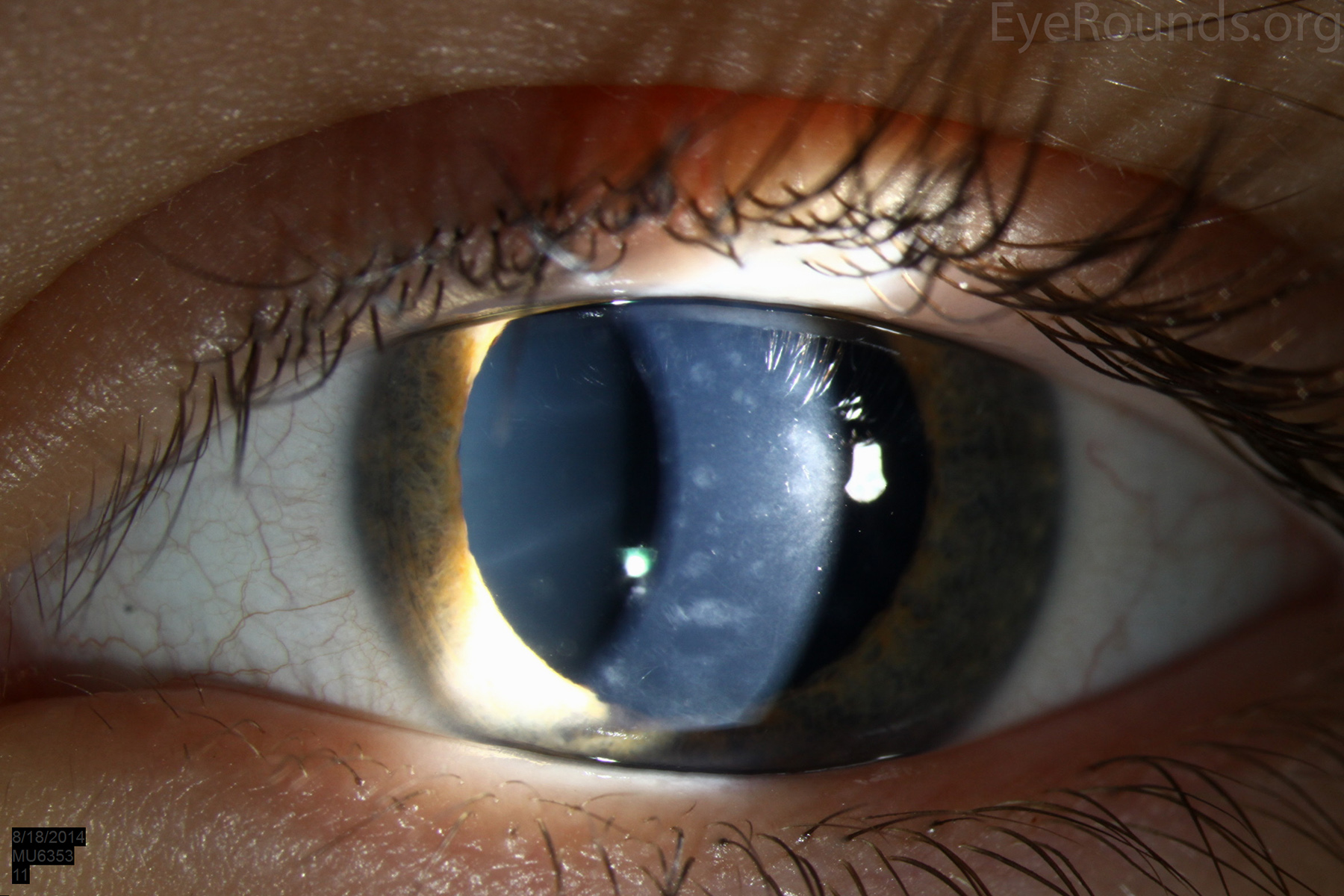
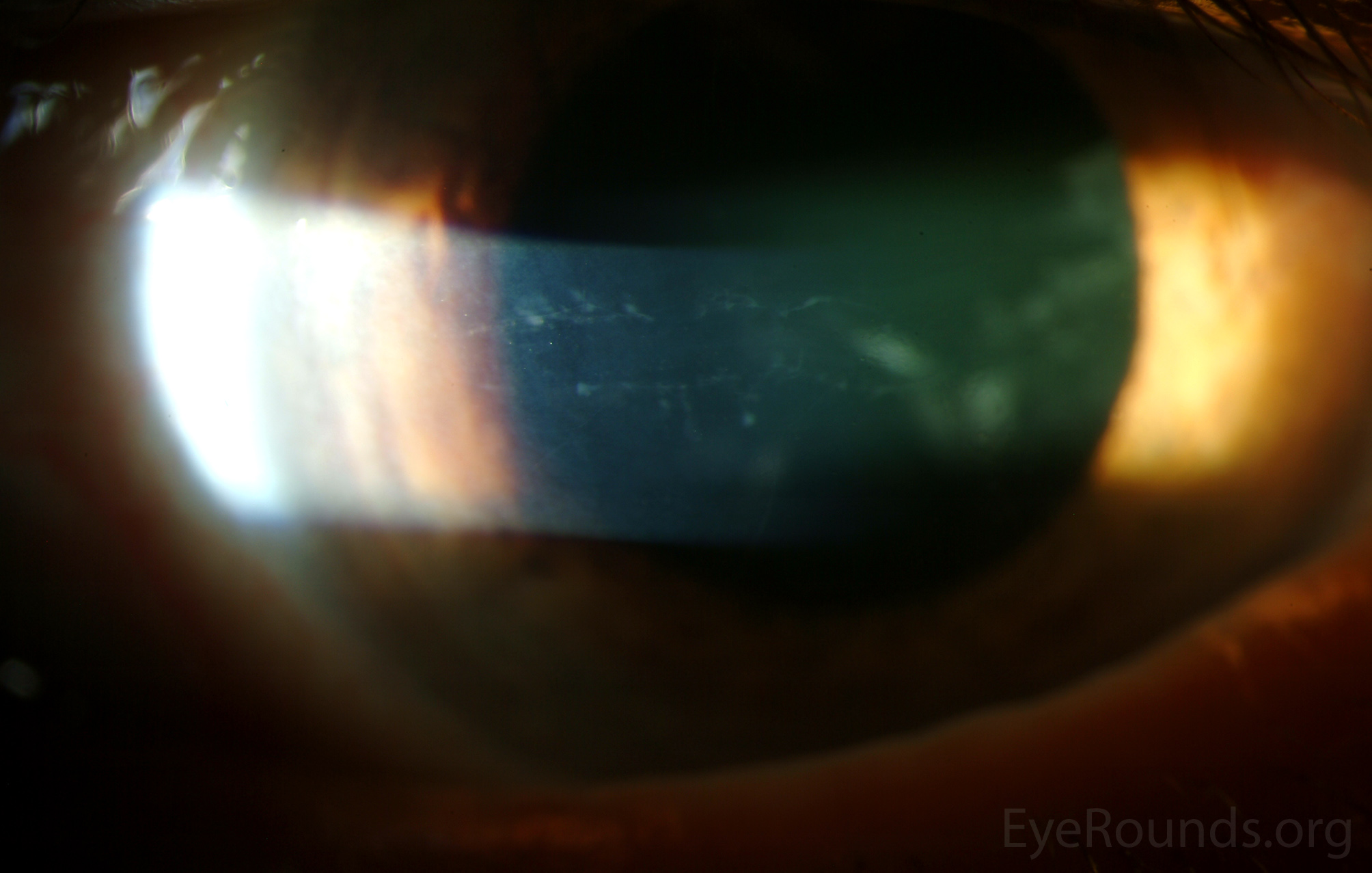
Diffusely scattered, well-circumscribed areas of stromal haze localized to the posterior 1/3 of the stroma. Most measure about 0.5 mm in size with intervening clear spaces. No associated neovascularization. No epithelial breakdown or erosions. (Fig. 1) |
Diffusely scattered, well-circumscribed areas of stromal haze localized to the posterior 1/3 of the stroma. Most measure about 0.5 mm in size with intervening clear spaces. No associated neovascularization. No epithelial breakdown or erosions. (Fig. 1) |
Anterior Chamber |
Deep and quiet |
Deep and quiet |
Iris |
Normal architecture, no iridocorneal adhesions or peripheral anterior synechiae (PAS) |
Normal architecture, no iridocorneal adhesions or peripheral anterior synechiae (PAS) |
Lens |
Clear |
Clear |
Vitreous |
Clear |
Clear |
Fundus |
Normal |
Normal |
Our patient had bilateral, diffuse, geographic-shaped, discrete, gray lesions at the level of Descemet's membrane (Fig. 1). His exam was most consistent with the diagnosis of posterior polymorphous corneal dystrophy (PPMD). With normal vision, absence of complaints, normal IOP, and absence of PAS or irido-corneal adhesions on slit lamp, there was no need to intervene and observation was recommended. The parents were examined and neither showed evidence of PPMD.
 |
 |
 |
Figure 1. Slit lamp photos of our patient showing diffuse, geographic-shaped, discrete, gray lesions at the level of Descemet's membrane as seen by direct illumination (a, b), and in a thin slit beam (c).
Posterior polymorphous corneal dystrophy (PPMD)
Posterior polymorphous corneal dystrophy (PPMD, PPCD), also known as Schlichting dystrophy, is a rare autosomal dominant disease that is a type of inherited corneal dystrophy [1] . Corneal dystrophies are typically bilateral, symmetric, slowly progressive, non-inflammatory, and without relationship to environmental or systemic factors. PPMD belongs to category 1 or 2 of the new ICD3 classification of corneal dystrophies [1] . Although it is autosomal dominant, the clinical expression and presentation of PPMD varies considerably even within the same family.PPMD can be asymptomatic and stable for many years, but it can also be progressive and debilitating. It usually presents in the second or third decade of life, with rare instances where it manifests earlier with corneal clouding [2].
The corneal abnormality in PPMD occurs at the level of Descemet's membrane and endothelium, and rarely will result in corneal edema and thickening. The three main patterns in which PPMD may present include endothelial vesicle-like lesions, band lesions, and diffuse opacities [2, 3].
 |
 |
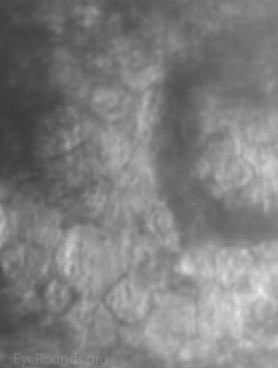 |
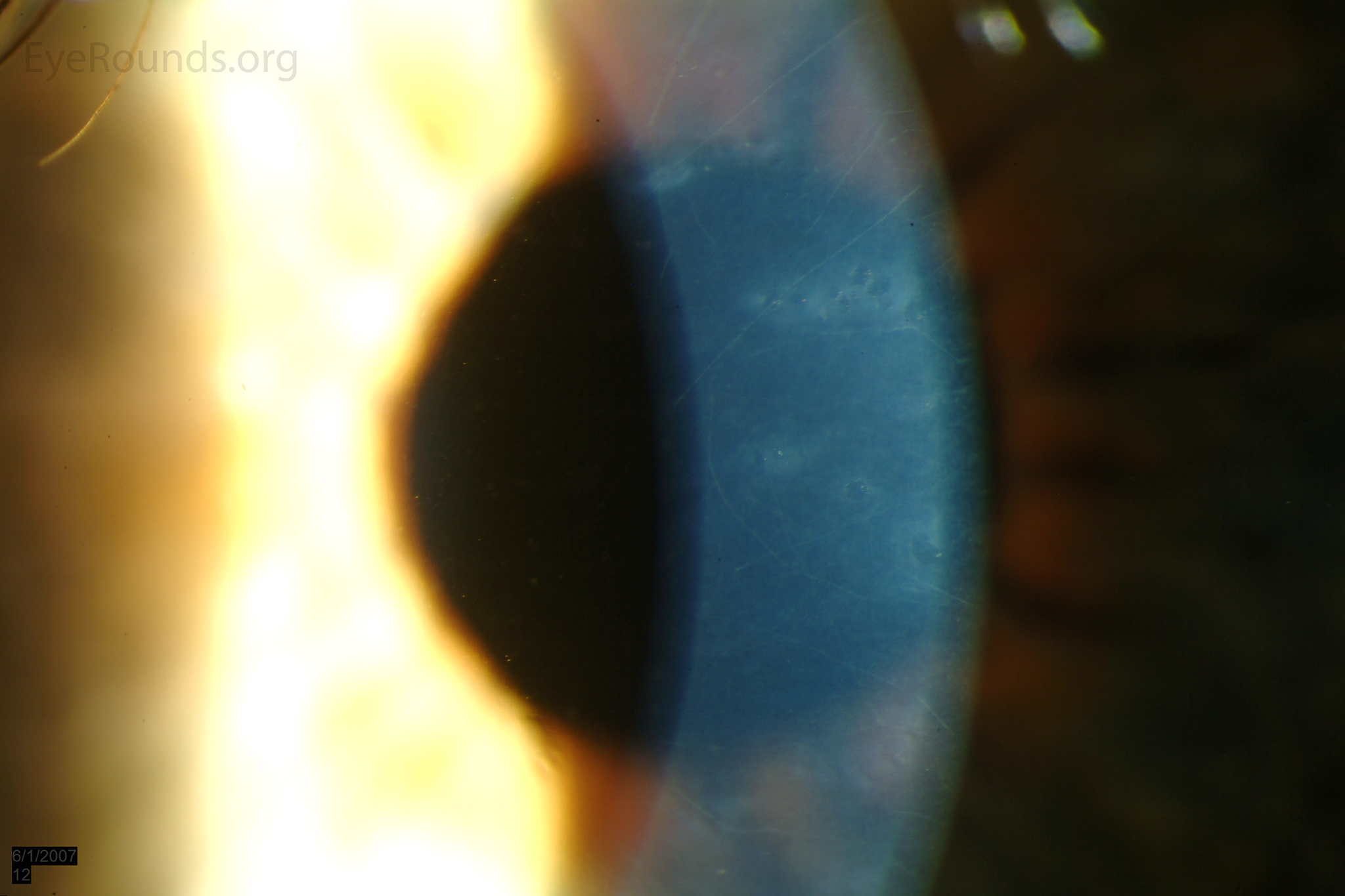
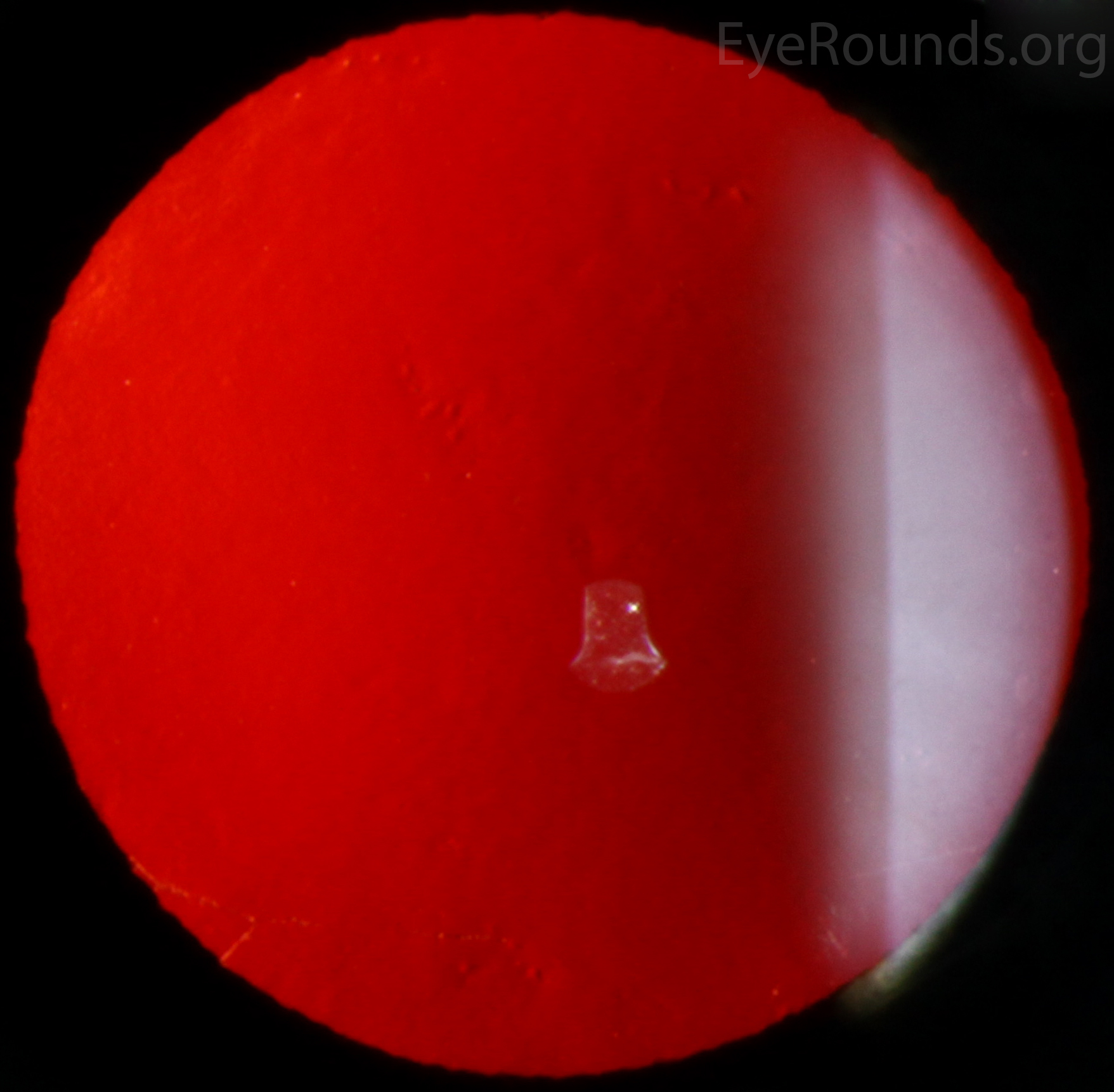
Figure 2. Vesicular lesions - the hallmark of PPMD- as seen on direct illumination (a), retroillumination (b). Vesicular lesions surrounded by normal endothelial cells as seen on specular microscopy (c).
The vesicular lesions are considered the hallmark of the disease and are present in almost all patients. They appear as circular or oval transparent cysts with a gray halo at the level of Descemet's membrane (Fig. 2a). They are best viewed by retroillumination with a widely dilated pupil when seen at the slit lamp (Fig. 2b). These vesicles are filled with fibrillar or collagenous material and are not fluid-filled spaces as the term 'vesicle' would imply [4]. On specular microscopy, these lesions, which range from 0.1 to 1 mm, appear as circular dark rings around a lighter center in which some cellular detail usually evident (Fig. 2c).
The band lesions can be seen on any area of the posterior cornea with the most frequent location just inferior to the central cornea [4]. These lesions, typically horizontal and ranging from 2 to 10 mm in length, have parallel scalloped edges and do not taper toward the ends in contrast to the tapered smooth-edged Descemet's tears seen in congenital glaucoma, trauma, and hydrops [5] (Fig. 3). On specular microscopy, these lesions demonstrate shallow trenches and ridges arising from a large number of confluent vesicles [6].
 |
 |
Figure 3. Scallop-edged endothelial band lesions as seen on direct illumination (a) and thin slit beam (b).
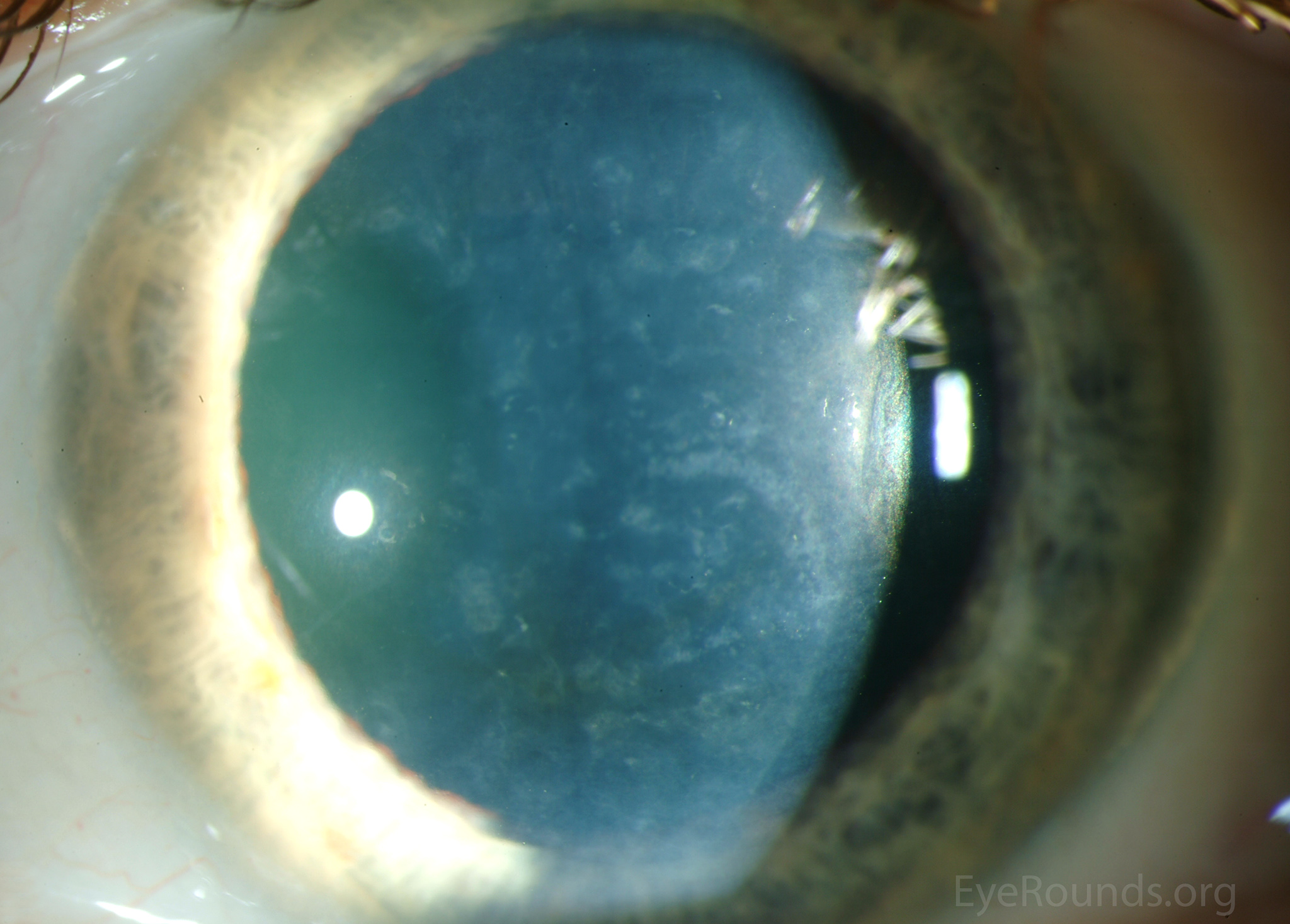
Diffuse opacities (the only lesions in our patient) are considered the least common feature, and appear as either small, macular, gray-white lesions or larger, sinuous geographic lesions at the level of Descemet's membrane. Direct illumination shows a haze in the posterior stroma adjacent to the lesions [2, 3] (Fig. 4). On retroillumination, the opacities show a peau d'orange texture. On specular microscopy the lesions appear as well-demarcated areas of enlarged and pleomorphic cells with indistinct borders and multiple reflective highlights surrounded by normal or small endothelial cells [4].
 |
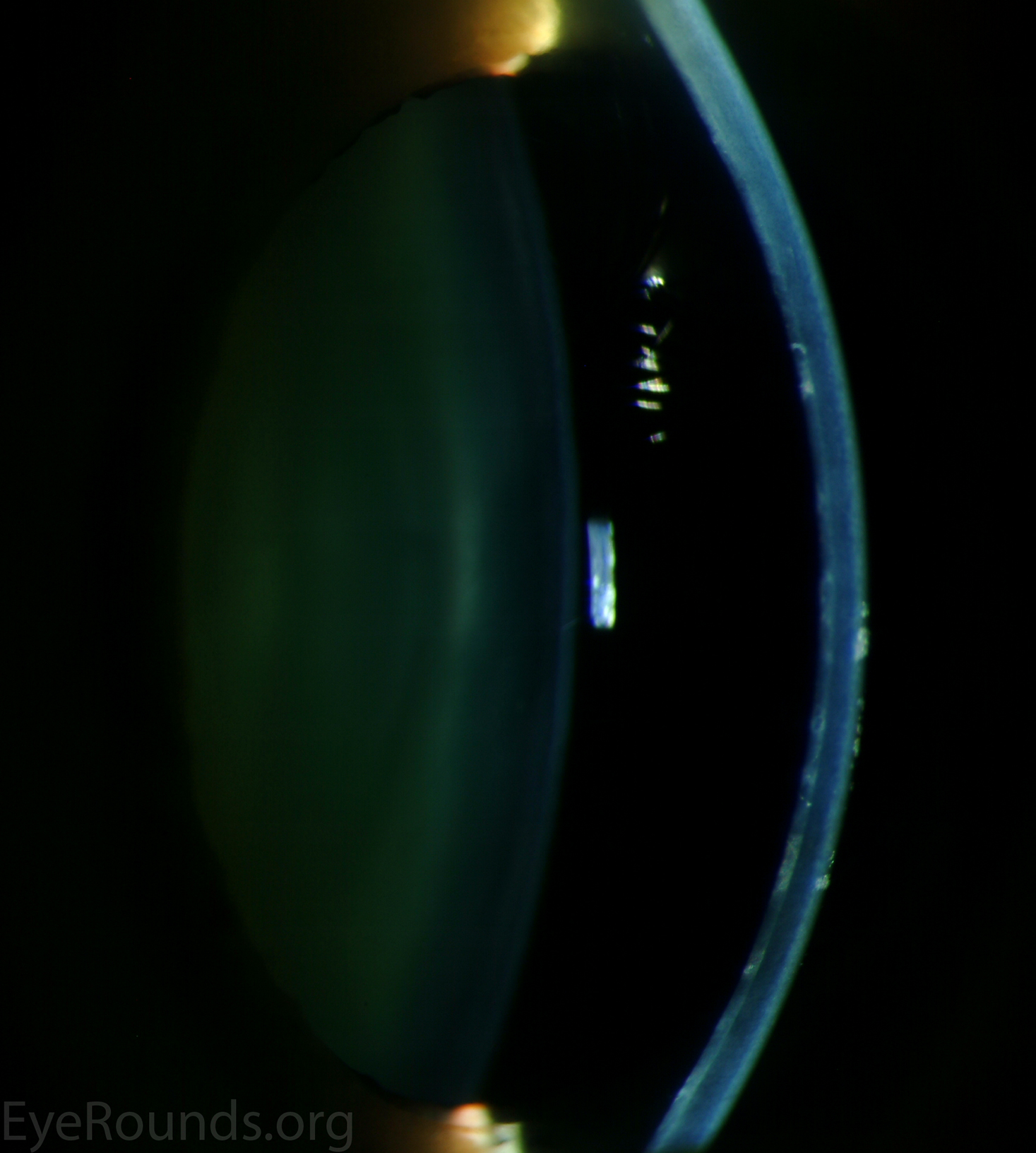 |
Figure 4. Diffuse inner corneal opacities as seen by direct illumination (a), and in a thin slit beam (b)
Patients presenting with the typical lesions of PPMD, including the diffuse opacities might exhibit broad-based irido-corneal adhesions and peripheral anterior synechiae (PAS) which usually result in elevation in IOP that is refractory to medical therapy and might require surgical interventions [2].
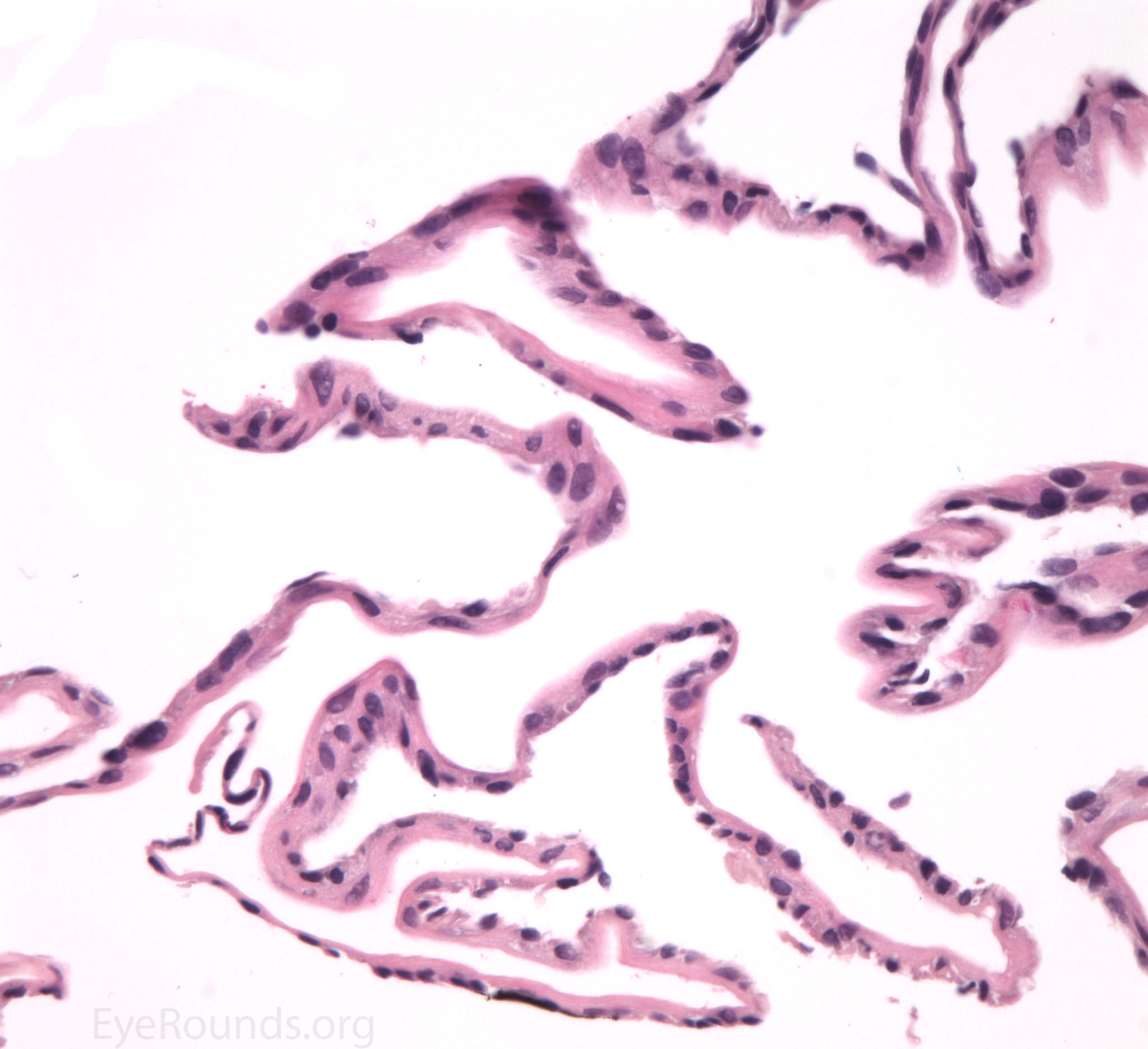
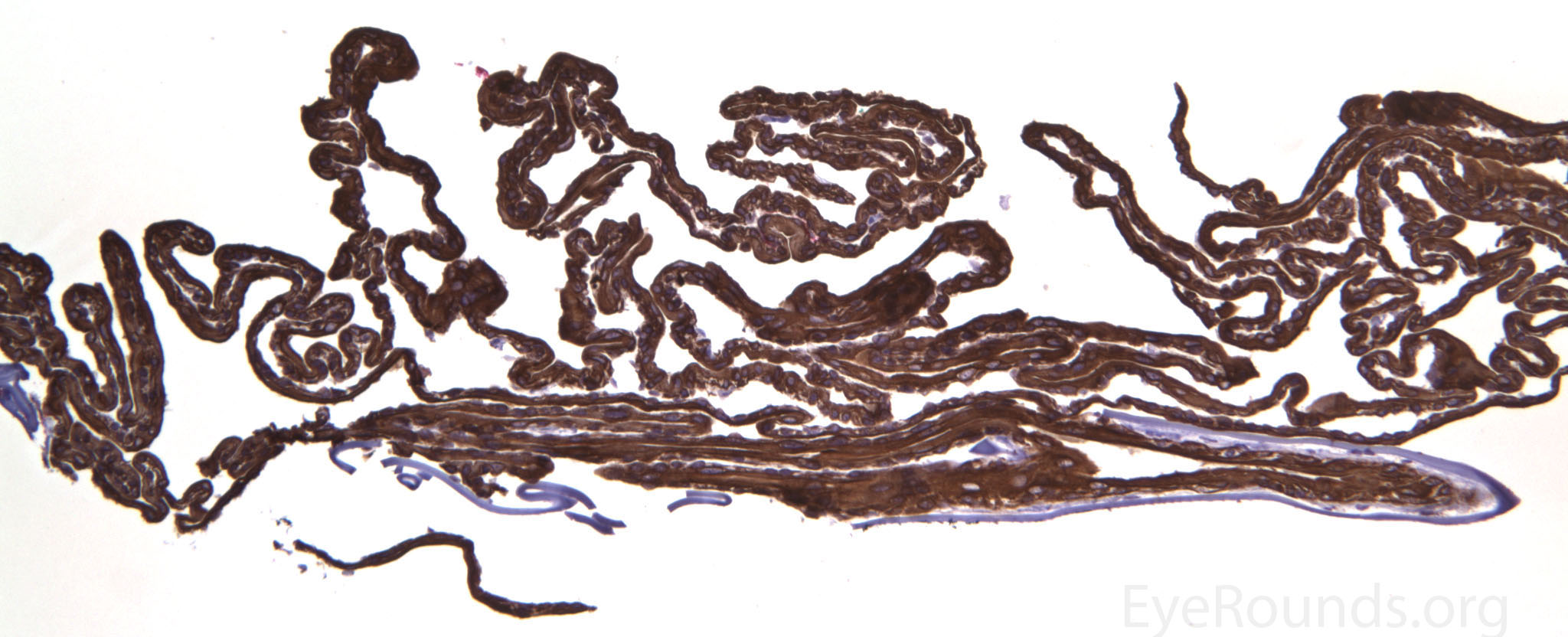
The pathogenesis of PPMD is attributed to an abnormal developmental differentiation of the endothelial cells [7]. Histologic studies have shown that the morphology of the endothelial cell layer resemble those of the epithelium (Fig. 5a). These multilayered epithelial-like cells are keratinized (Fig. 5b, c), connected by well-developed desmosomes, and appear to have microvilli when examined under scanning electron microscopy [7, 8]. Moreover, these cells can migrate over the trabecular meshwork and iris, causing extensive peripheral synechiae and glaucoma [9].
Several genome-wide linkage analyses on different families with PPMD have identified several gene loci responsible for this hereditary disease. These include the long arm of chromosome 20 (20p11.2-q11.2) [10] designated as PPMD 1, the short arm of chromosome 1 (1p34.3-p32) [11] designated as PPMD 2, and the short arm of chromosome 10 (10p11.2) [12] now designated as PPMD 3.
 |
 |
 |
|
 |
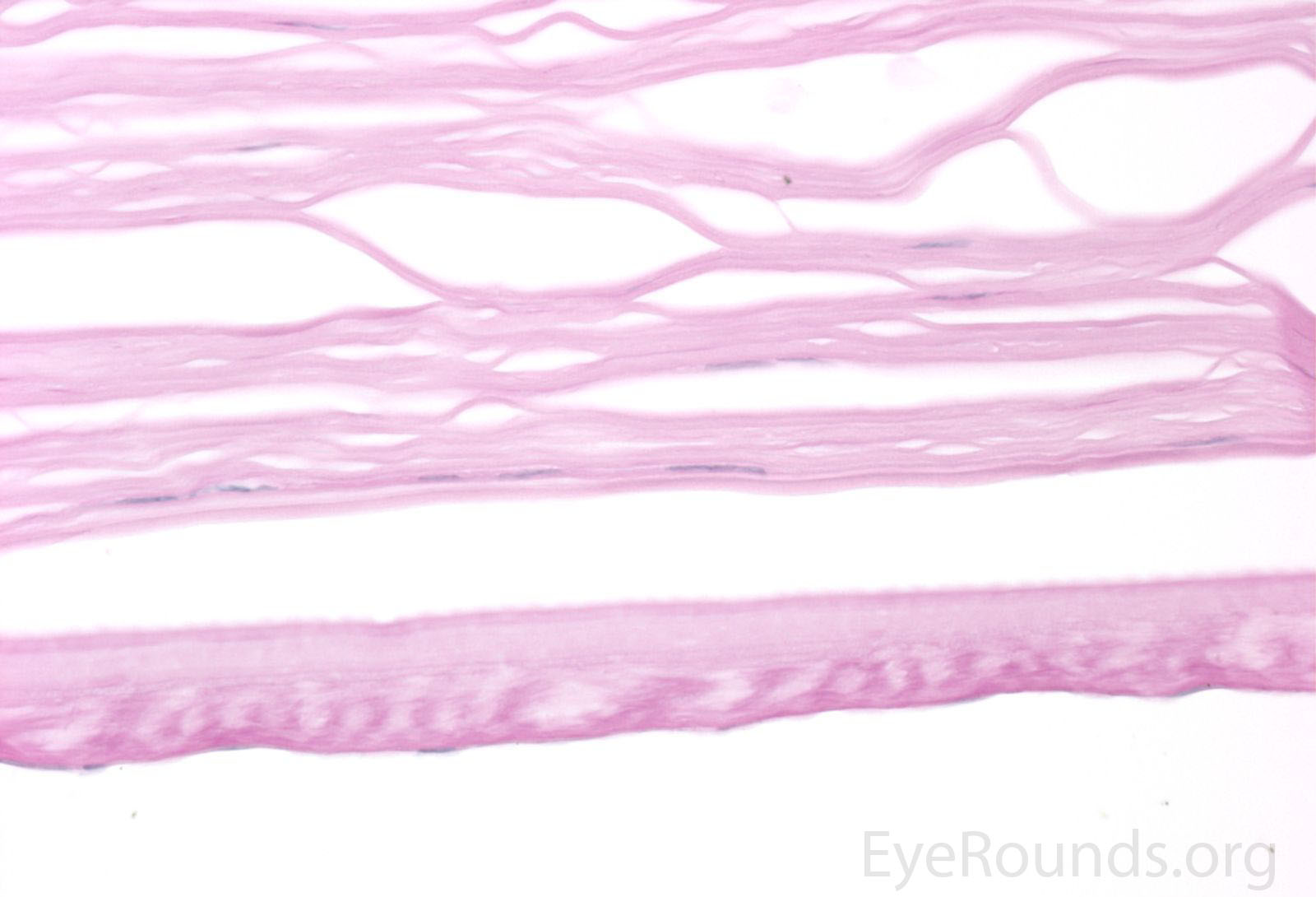 |
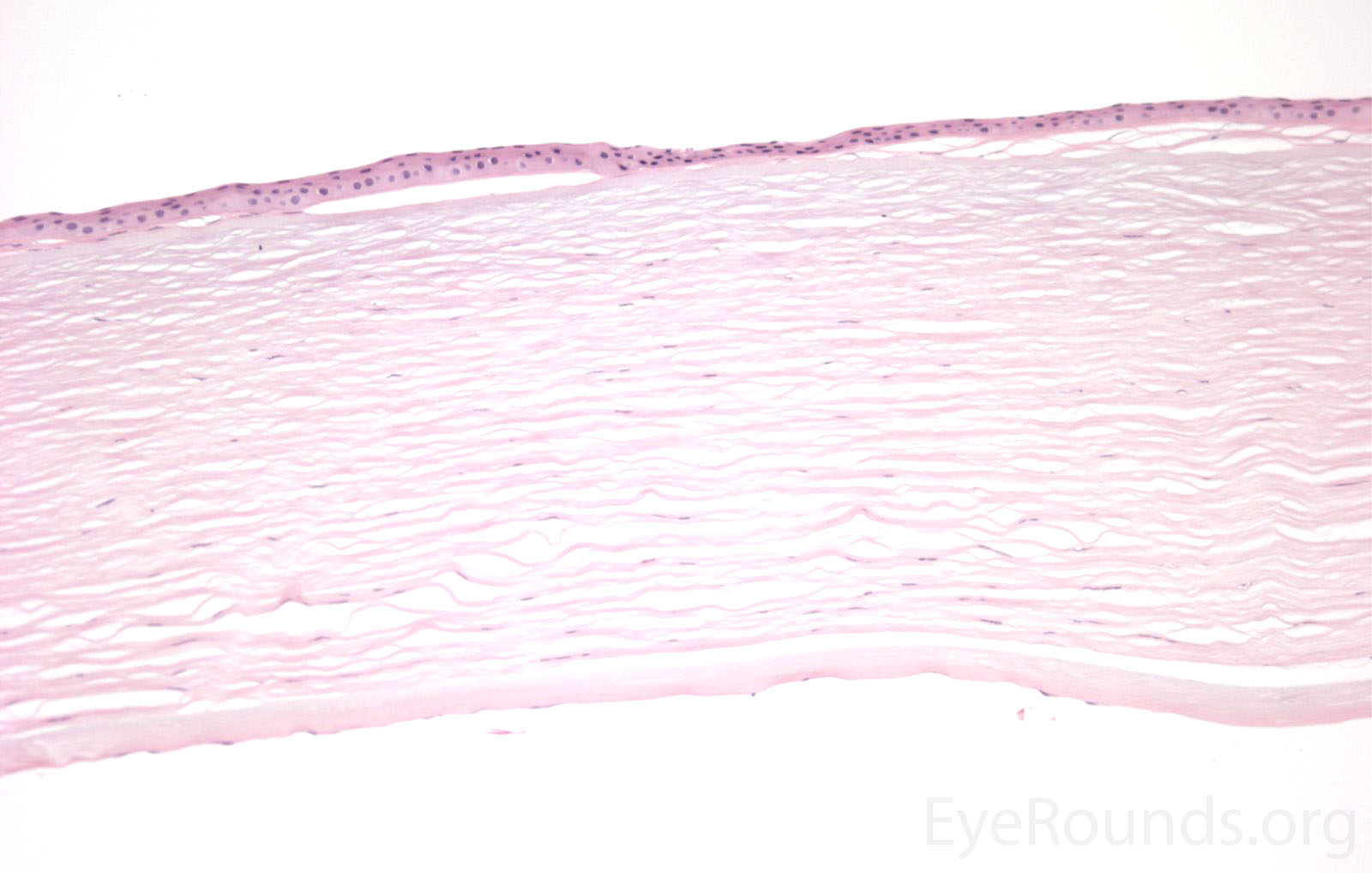
Figure 5. Endothelial cell metaplasia in PPMD as seen with H&E stain (a). Positive staining with AE1/AE3 (b) and CK5-6 (c) which stains cytokeratin, an epithelial cell marker (a,b,c represents Descemet membrane and endothelial cell layer obtained from a PPMD patient during endothelial keratoplasty). H&E stain of full-thickness corneal tissue obtained from PPMD patient showing bullous keratopathy with loss of endothelial cells, loss of artifactitious clefting in the stroma, subepithelial bullae, and hydropic degeneration as well as a thickened, acellular layer between DM and the endothelium, which can be seen in PPMD (d-50X, e-200X).
Most patients with PPMD are asymptomatic and are diagnosed on routine eye exam. These patients usually have a great prognosis and often do not require any treatment. In a small number of cases however, the disease may be extensive and progressive resulting in corneal edema, visual disturbance, and increased intraocular pressure necessitating surgical intervention. Mild corneal edema can be managed with sodium chloride 5% drops and ointment, a soft bandage contact lens for ruptured bullae, and stromal micropuncture for areas of localized swelling and poor endothelial adherence.
For severe cases, the gold-standard surgical therapy was previously penetrating keratoplasty. In the largest series of PPMD cases reported (120 individuals), thirteen patients required corneal transplantation [2]. Fifty percent of patients undergoing transplantation attained 20/40 vision postoperatively and 55% of patients maintained clear grafts. In this series, 14% of the patients developed an increase in IOP, and this was mainly a result of irido-corneal adhesions that closed portions of the filtration angle. The increase in IOP should be initially managed with medications that decrease aqueous production. The use of miotics may be helpful if areas of open angle are present. Laser trabeculoplasty is not recommended in these patients since it might accelerate synechiae formation and aggravate the situation. In those patients whose intraocular pressure cannot be controlled medically, filtering surgery is probably the safest option.
The prognosis for surgery correlated with the presence of PAS and elevated intraocular pressure. Eighty percent of patients with PAS visible on slit lamp exam without gonioscopy had postoperative vision worse than 20/400. Sixty nine percent of patients with increased intraocular pressure preoperatively had postoperative vision worse than 20/400.
This being said, the study concluded that the presence of PAS visible without gonioscopy and increased intraocular pressure must be considered relative contraindications to corneal transplantation in PPMD [2]. This study also noticed recurrence of PPMD in 4 of the 22 corneal transplants. This recurrence appears to be caused by the overgrowth of the original diseased host endothelium [13].
Recently, several case reports have shown success in using endothelial keratoplasty in earlier cases without significant stromal opacification (DSAEK in an 8-month-old patient [14], DMEK-S in a 20-year-old patient [15], and DSEK in a 12-year-old patient [16]). In our personal experience, we have had success with the DMEK procedure in individuals with PPMD and this is currently our procedure of choice in many patients with this condition. Although these results are promising, long-term follow-up is needed, and studies comparing the recurrence rate of PPMD in endothelial keratoplasty compared to penetrating keratoplasty are still needed.
EpidemiologyRare autosomal-dominant disorder with variable expressivity Symptomatic patients typically present between age 25-50 years More severe disease can present as corneal opacification in the first decade of life May be associated with Alport syndrome |
SignsCorneal opacification that is typically bilateral, but often asymmetric Changes occurs at the level of Descemet's membrane and endothelium and include vesicle-like lesions (Fig. 2), band lesions (Fig. 3), and diffuse opacities (Fig. 4) Associated features include iridocorneal adhesions, peripheral anterior synechiae which usually indicate more aggressive disease
|
PathophysiologyFocal metaplasia of endothelial cells into a population of aberrant, keratinized epithelial-like cells Genetic lociPPMD 1 — 20p11.2–q11.2 - gene unknown PPMD 2 — 1p34.3–p32.3 - collagen type VIII alpha 2 - COL8A2 PPMD 3 — 10p11.2 - two-handed zinc-finger homeodomain transcription factor 8—ZEB1 |
TreatmentThe majority of patients have no symptoms thus require no treatment; those with corneal opacification and visual disturbance are offered keratoplasty Regular IOP monitoring is needed. Increase in IOP should be managed with drugs that decrease aqueous production: β blockers, α-adrenergic agonists, and carbonic anhydrase inhibitors. When medical management fails, the next step should be filtering surgical interventions
|
Dahrouj M, Vislisel JM, Raecker M, Maltry AC, Goins KM. Posterior Polymorphous Corneal Dystrophy (PPMD). Feb 23, 2015; Available from: https://eyerounds.org/cases/208-PPMD.htm

Ophthalmic Atlas Images by EyeRounds.org, The University of Iowa are licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License.

Address
University of IowaLegal
Related Links
