Authors: Ryan J. Diel MD, Tirth Shah MD, Randy Kardon MD, Michael Wall MD
September 17, 2020
INITIAL PRESENTATION
Chief Complaint: Progressive vision loss left > right
History of Present Illness:
A 9-year old female was referred to the University of Iowa to undergo additional evaluation for profound vision loss with “optic disc swelling.” The child presented with her mother and related progressive blurry vision of both eyes that began 4 days prior to evaluation. The child’s mother stated that her daughter began having difficulty distinguishing colors prior to the onset of her vision loss and described colors as appearing “dim.” The child also endorsed mild achiness of the left eye with subsequent achiness of the right eye both associated with the onset of blurry vision. Additionally, headaches were present which worsened when lying down. She denied nausea, vomiting, ringing in her ears, diplopia, or episodes of transient vision loss
The patient’s mom reported no cats or other animals in the household, no recent travel, no recent outdoor activities such as hiking, and no recent reported tick bites.
Past Ocular History: None
Medical History: None
Past Surgical History: None
Medications:
None
Allergies: No known drug allergies
Family History: :
Father – glaucoma, astigmatism
Great aunt – possible similar symptoms at a young age
Social History:
Starting 4th grade
No notable stressors at home or at school
Review of Systems:
Treated urinary tract infection several weeks ago. No other recent illnesses or changes in her health were reported by mom.
OCULAR EXAMINATION
Visual Acuity
Right eye (OD): Count fingers at 1’
Left eye (OS): Hand motion at 1’
Ocular Motility/Alignment
Full versions in both eyes (OU) but painful
Intraocular Pressure (by Tonopen)
OD: 13 mmHg
OS: 17 mmHg
Pupils
OD: 7.5mm in dim light, constricting to 6.5mm with bright light; minimally reactive secondary to pharmacologic dilation prior to arrival
OS: 7.5mm in dim light, constricting to 6.5mm with bright light; minimally reactive secondary to pharmacologic dilation prior to arrival
Confrontational Visual Fields (count fingers)
Deferred in the setting of poor visual acuity
External
OU: normal
Slit Lamp Exam
Lids/lashes: Normal OU
Conjunctiva/sclera: Clear and quiet OU
Cornea: Clear OU
Anterior Chamber: Deep and quiet OU
Iris: Normal architecture, pharmacologically dilated OU
Lens: Normal OU
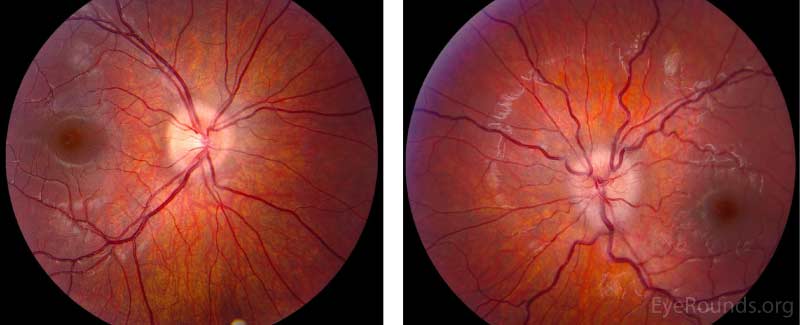
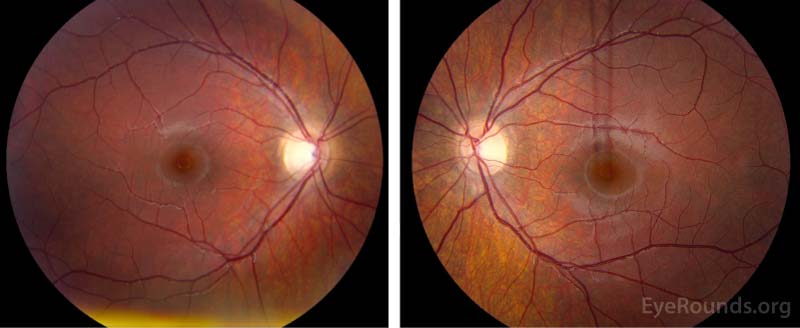
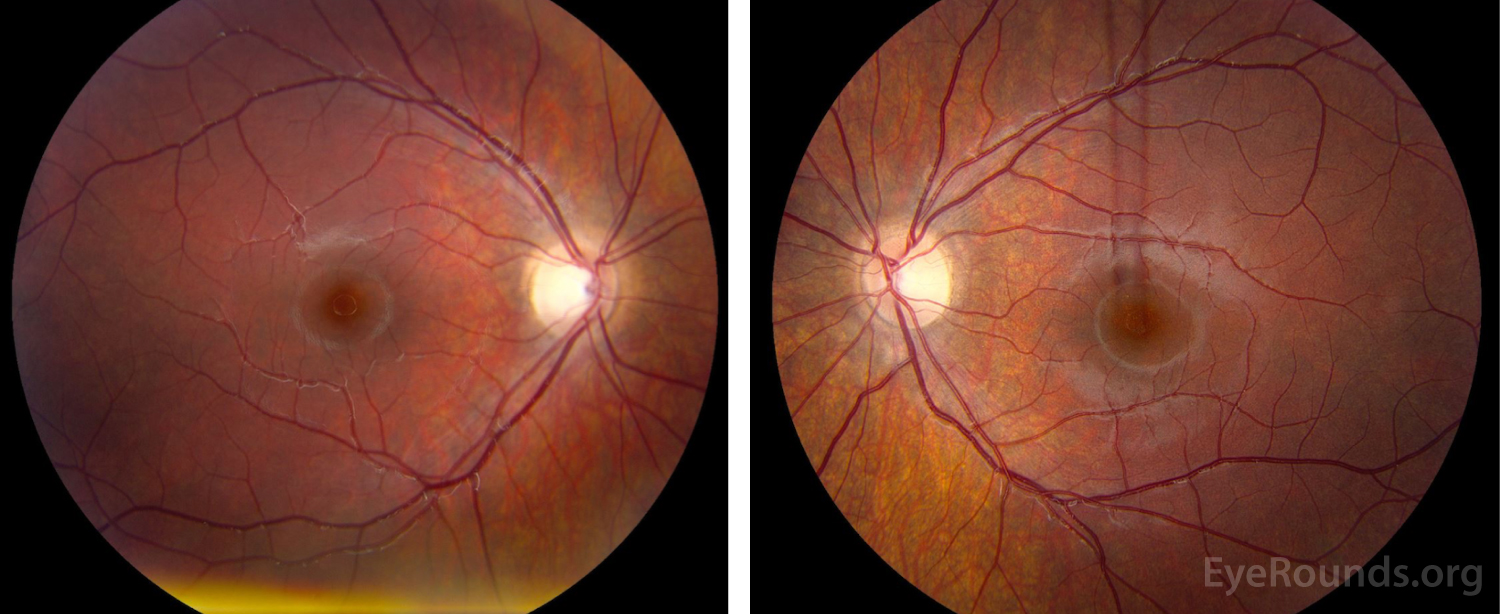
Dilated Fundus Examination (Figure 1):
Vitreous: Normal OU
Disc:
OD: Grade I disc edema, no pallor, no hemorrhages, no exudates
OS: Grade II disc edema, no pallor, no hemorrhages, no exudates
Cup-to-disc: no cup noted in either optic nerve
Macula: Normal OU
Vessels: Normal OU
Periphery: Normal OU
Differential Diagnosis
Idiopathic intracranial hypertension
Bilateral optic neuritis
Infiltration of the optic disc
Intracranial mass with obstructive hydrocephalus and compression of anterior visual pathway
Additional Testing
Imaging
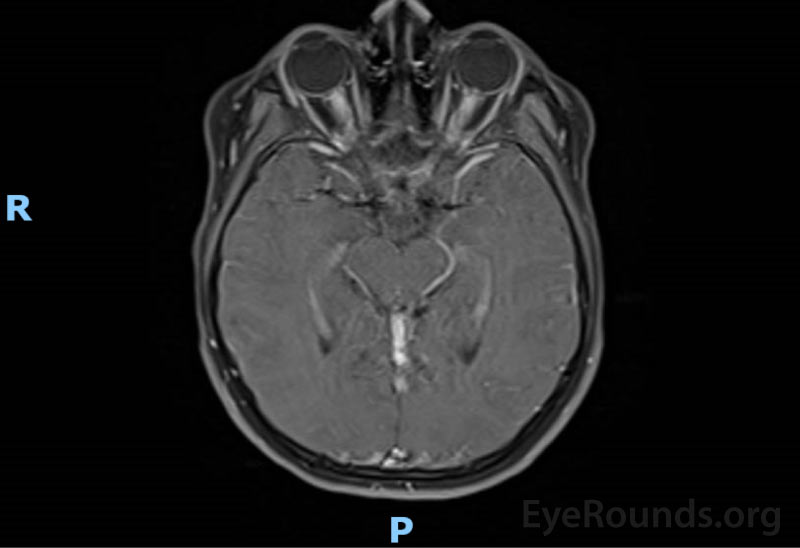
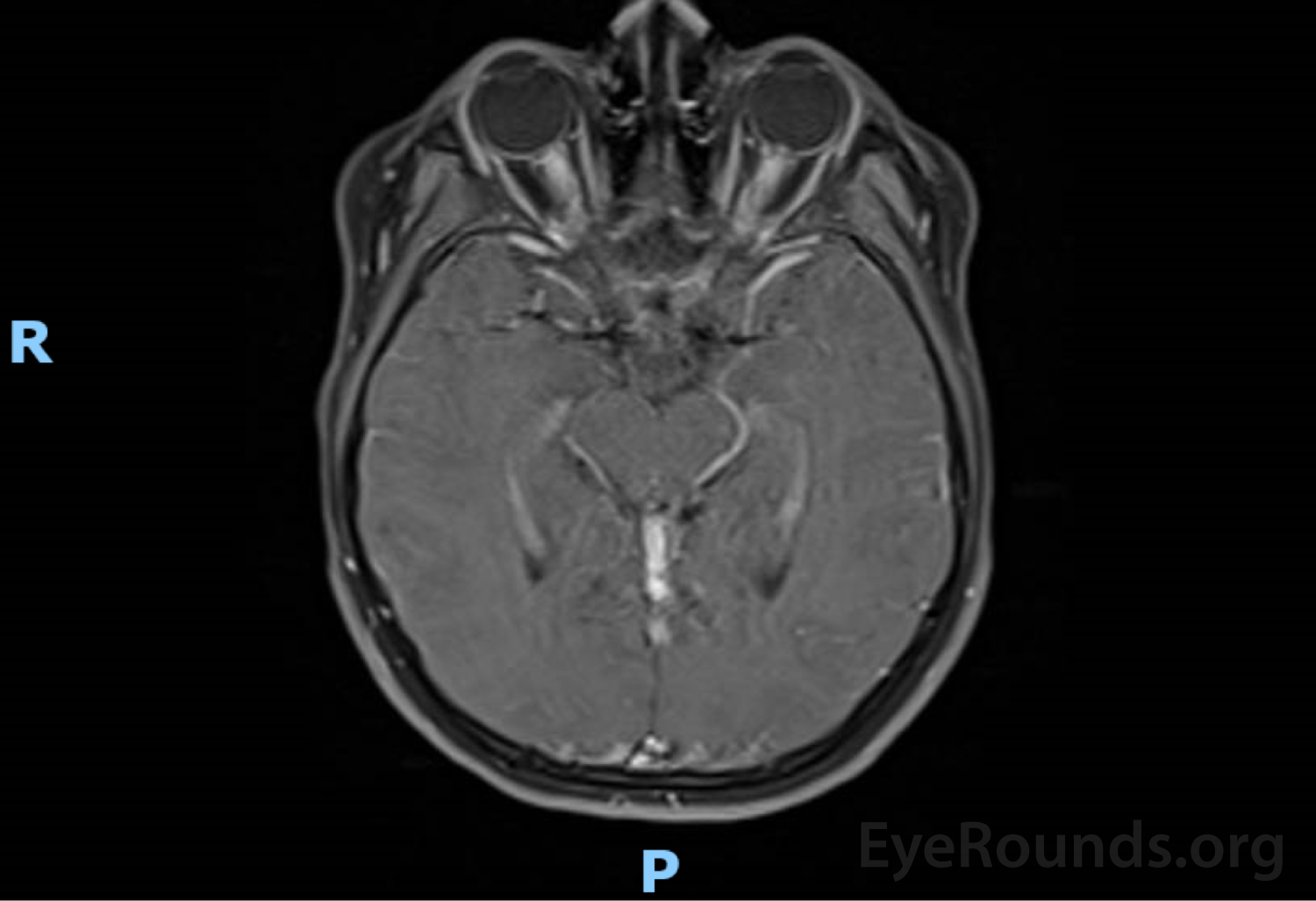
(Figure 2) MRI brain and orbits w/ and w/o contrast: enhancement and enlargement of intraorbital segments of optic nerves, left > right
MRI cervical, thoracic, lumbar spine: normal
Labs
CBC w/ differential: normal
ESR: 19 mm/Hr (ref 0-20 mm/Hr)
Anti-nuclear antibody <1:80
Anti-double stranded DNA qualitative: indeterminant
Anti-double stranded DNA quantitative: 6 (ref 0-4 IU/mL)
Anti-neutrophil cytoplasmic antibody: negative
Bartonella henselae IgG/IgM titer: negative
Lyme IgG/IgM titer: negative
Lumbar puncture w/ cytology and opening pressure:
Pink, hazy, no xanthochromia, total protein: 22, glucose: 73, RBC: 1000, total nucleated cells: 8 (0 neutrophils, 7 lymphocytes), opening pressure 26cm
Meningitis/encephalitis panel: negative for bacterial and viral agents
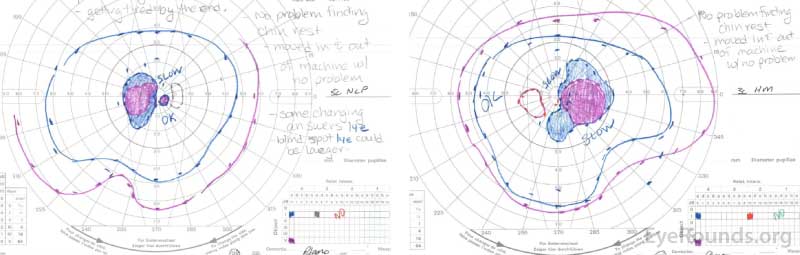
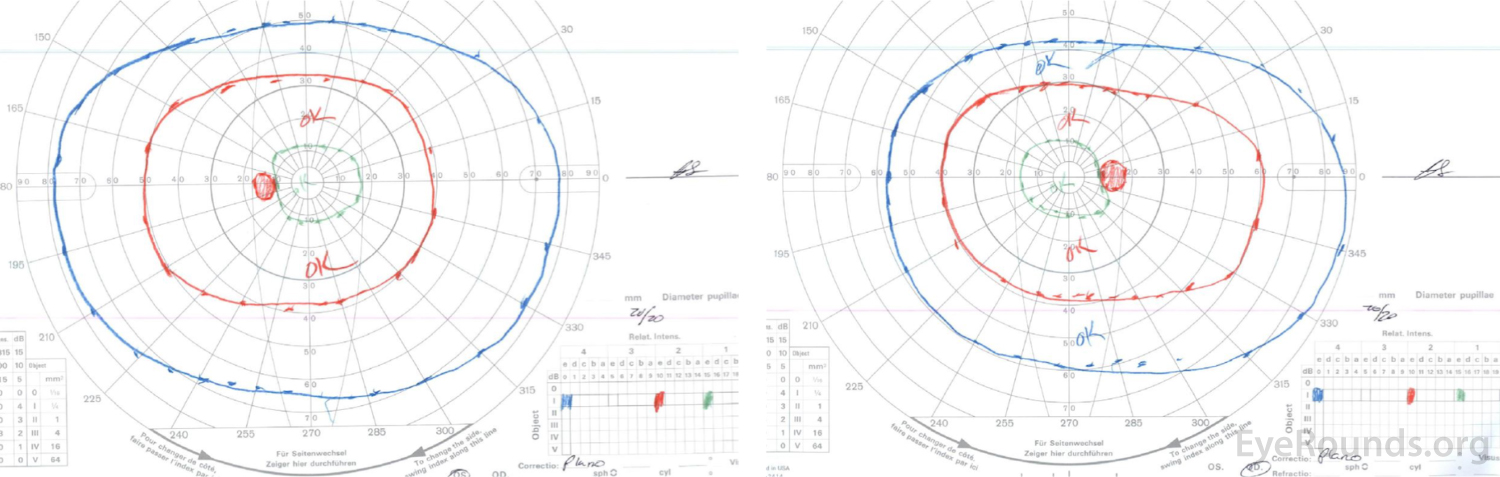
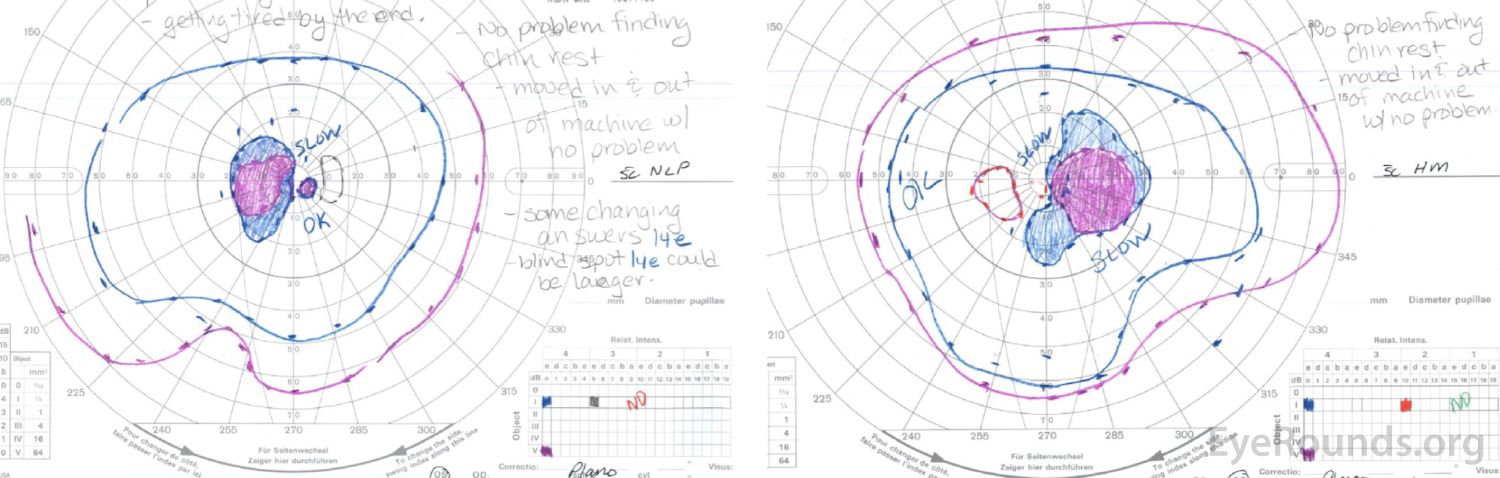
Right eye (right image) demonstrates complete loss of the I1e isopter, marked constriction of the I2e with only a small island remaining nasally, and a dense cecocentral scotoma with complete loss of the V4e.
Left eye (left image) demonstrates complete loss of the I2e isopter, marked constriction of the I3e with small island remaining nasally, and a dense cecocentral scotoma with complete loss of the V4e.
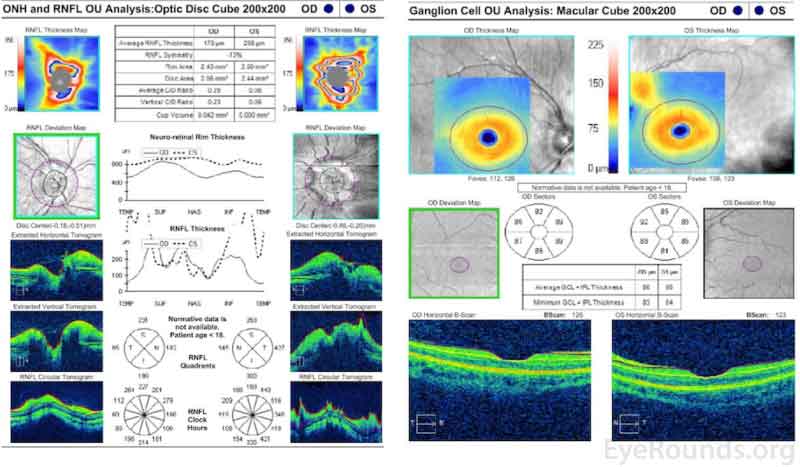
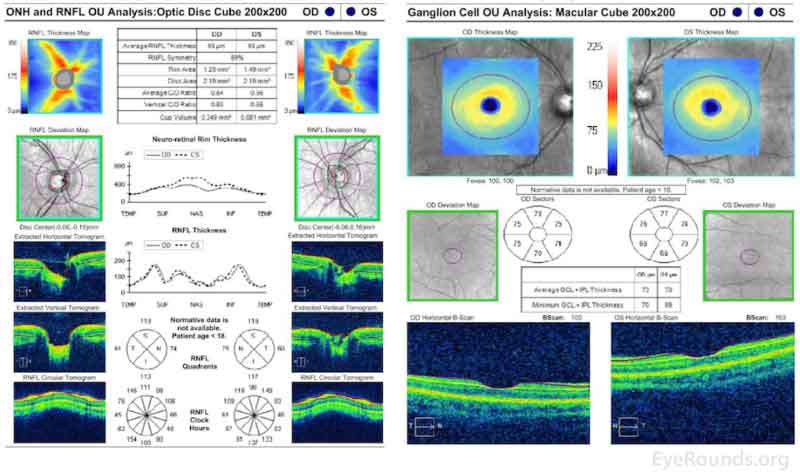
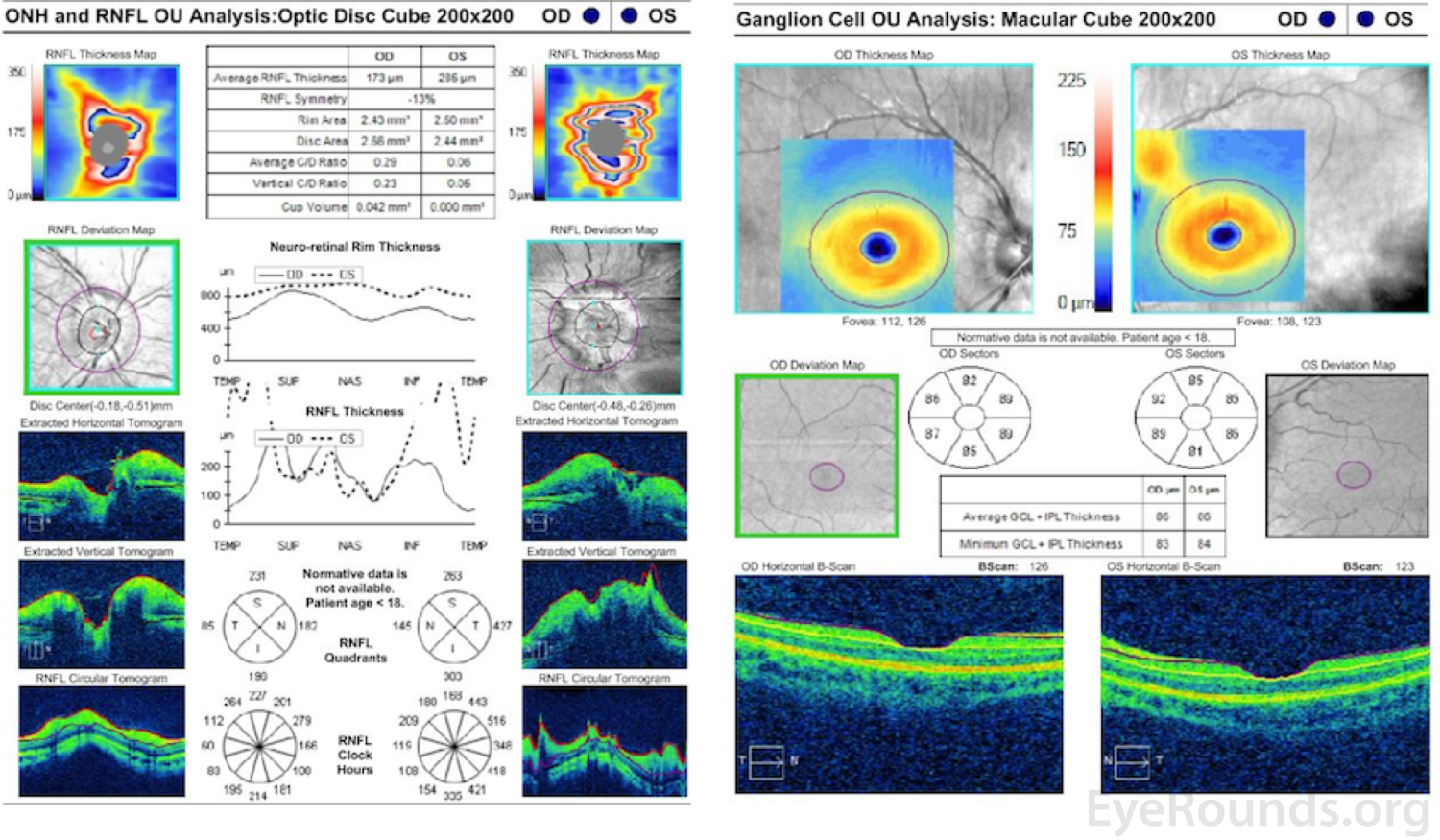
Optical Coherence Tomography (Figure 4)
Marked elevation and distortion of the optic nerve head leading to artifactual thickening of the retinal nerve fiber layer and ganglion cell layer OU.
DIAGNOSIS: MOG-associated optic neuritis
Figure 1. Dilated Fundus Examination. Vitreous: Normal OU.
Disc:
OD: Grade I disc edema, no pallor, no hemorrhages, no exudates,
OS: Grade II disc edema, no pallor, no hemorrhages, no exudates.
Cup-to-disc: no cup noted in either optic nerve.
Macula: Normal OU.
Vessels: Normal OU.
Periphery: Normal OU.
Right eye (right image) demonstrates complete loss of the I1e isopter, marked constriction of the I2e with only a small island remaining nasally, and a dense cecocentral scotoma with complete loss of the V4e.
Left eye (left image) demonstrates complete loss of the I2e isopter, marked constriction of the I3e with small island remaining nasally, and a dense cecocentral scotoma with complete loss of the V4e.
Figure 4. Optical Coherence Tomography. Marked elevation and distortion of the optic nerve head leading to artifactual thickening of the retinal nerve fiber layer and ganglion cell layer OU.
The MOG antibody testing took several days to be resulted; however, given the clinical diagnosis of bilateral optic neuritis due to observed optic nerve enhancement on MRI, optic disc edema, and profound vision loss, the patient was admitted to the pediatric neurology service for emergent IV methylprednisolone therapy. After 2 doses of IV steroids, her vision improved to 20/250 OD and 20/400 OS. Prior to being discharged, she received a total of 3 days of IV steroids and continued 60mg oral prednisone as an outpatient.
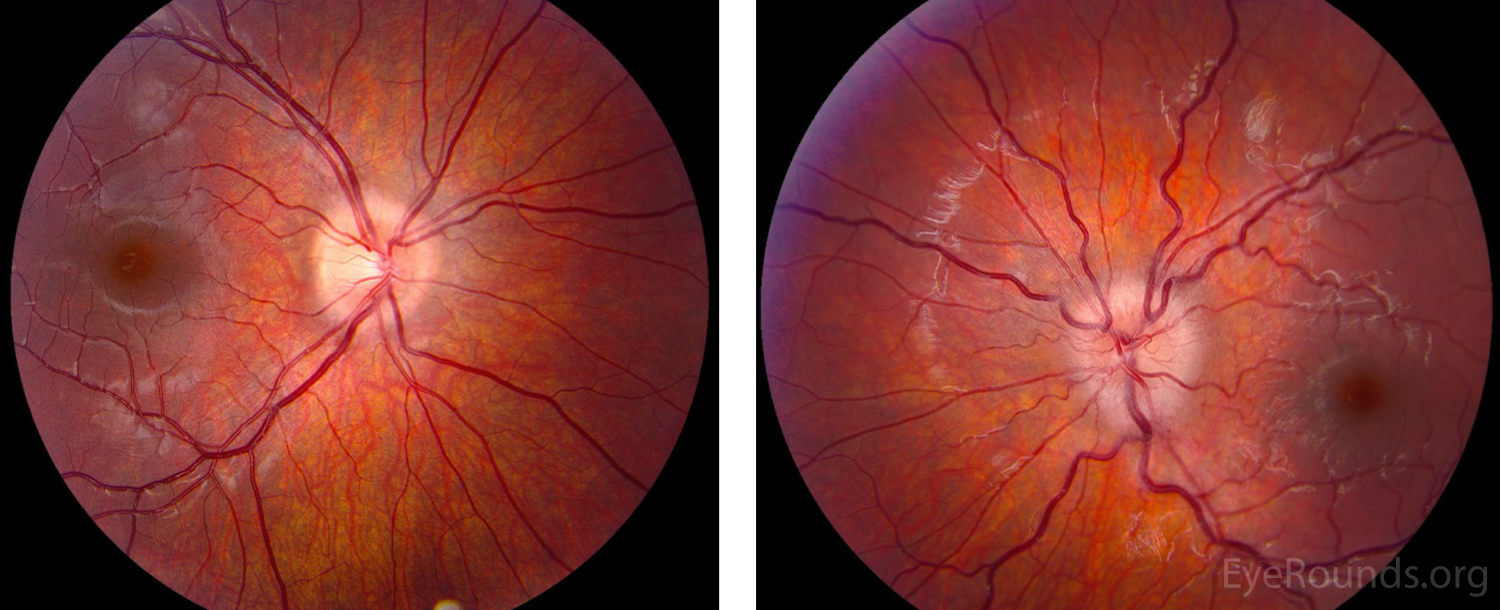
At her 1-month follow-up visit, the patient’s vision had returned to 20/20 OU, her disc edema had resolved (Figure 5), her repeat Goldmann perimetry showed full isopters (Figure 6), and OCT of her optic nerve also normalized (Figure 7). The patient has since been tapered off of steroids without any recurrent episodes at the time of this article.
Biomarkers have changed the paradigm for characterizing and correctly diagnosing CNS inflammatory and demyelinating diseases. Myelin oligodendrocyte glycoprotein (MOG)-IgG was initially thought of as a marker of multiple sclerosis [1] and acute disseminated encephalomyelitis (ADEM) in children [2]. However, recent studies have found optic neuritis (ON) to be its predominant phenotype in both the pediatric and adult populations. Most importantly, recent advances have categorized MOG-IgG associated disorders (MOGADs) as distict from both multiple sclerosis and aquaporin-4 (AQP4)-IgG-positive associated disorders. Thus, despite the clinical overlap between these three entities, it is crucial to appreciate that they are separate diseases with differing pathophysiology, clinical course, and most importantly, different treatments.
Etiology/Epidemiology:
MOG-IgG positive optic neuritis is generally seen in children or young adults, classically peaking around the third decade with an equal preponderance between females and males.[1 ,3] In contrast, multiple sclerosis ON and AQP4-IgG positive ON are generally seen in females in their second decade and fourth decade, respectively.[1] Most studies have found that among patients with ON, the MOG antibody is detected in approximately 15% of patients, which is more frequent than that of AQP4 antibody, which is seen in 2% of cases.[4] While the majority of patients will present with positive antibody titers, the antibody titer may fluctuate resulting in testing becoming negative then turning positive again.[5] Among children with ON, MOG+ is more common in teenagers (13-18 years), whereas encephalitis is more predominant in the younger age group (4-8 years).[6 ,7] Recent cohort studies have reported positive MOG antibodies in 46% of pediatric optic neuritis cases,[8] suggesting this antibody is more prevalent than originally presumed. The proportion of MOG positive antibodies surpasses that of AQP4 positive antibody associated optic neuritis in each cohort, and those children with MOG positive antibodies tended to be younger.[8-10] Importantly, both a monophasic and recurrent disease course may be seen, although overall a recurrent disease phenotype is more common.[11]
Pathophysiology:
Our current understanding of the pathophysiology behind MOG-IgG associated disorders is still evolving. From a cellular perspective, we know that MOG is a transmembrane protein that forms a minor component of the CNS myelin sheath. MOG serves as an adhesion molecule and helps maintain stability of oligodendrocytes.[12] However, it is also highly immunogenic.[13] Unlike AQP4-Ab, which is known to cause destruction of astrocytes,[14] MOG-Ab is thought to cause inflammation and myelin destruction without directly affecting astrocytes[15 ,16]. The preservation of astrocytes seen in MOG-IgG seropositive groups is the most remarkable difference in the pathophysiology between these two entities and perhaps may explain the more mild disease course seen MOG-AD in comparison to AQP4 antibody associated disorders.[17]
Signs/Symptoms:
Patients typically present with severe loss with accompanying bilateral optic disc edema.[11] According to a 2018 study of 246 patients, the median nadir of visual acuity during an optic neuritis attack were 20/100 in multiple sclerosis ON, hand motion in AQP4-IgG ON, and count fingers (logMAR 1.6) in MOG-IgG ON.[3] The median visual acuity at last follow up (~6 months) was 20/25 in multiple sclerosis ON, count fingers in AQP4-IgG ON, and 20/25 in MOG-IgG ON. Thus, the recovery and final visual outcomes following immunosuppressive treatment is typically significantly better than that seen in AQP4-IgG positive ON.[3 ,11] .
In contrast to AQP4-IgG ON and multiple sclerosis ON, relapses are generally more frequent although shorter in duration in patients with MOG-IgG ON.[18 ,19]
Clinically, it’s important to recognize that a patient presenting with optic neuritis and significant disc edema and/or visual acuity less than 20/200 should likely prompt further investigation for serum MOG and AQP4 antibodies. Clinically significant optic disc edema is generally not a prominent feature in multiple sclerosis ON whereas it is fairly common in MOG-IgG ON. [4]. Optic disc edema is especially important to identify during the initial presentation since this feature could be absent due to atrophy from recurrent ON during subsequent evaluations. Based on a multicenter, prospective study involving sixty-five patients conducted by Ducloyer et al., the authors advocated the testing for MOG antibody only in the case of either optic disc edema, bilateral, or recurrent ON.[4] Other clinicians feel it is important to test serum MOG-IgG and AQP4-IgG in cases where visual acuity is less than 20/200 or when radiological/CSF findings are inconsistent with multiple sclerosis ON.[1] A comprehensive list of signs that could prompt testing for serum MOG-IgG or AQP4-IgG can be found in Table 2.
Table 1: Comparison between MOG-IgG, AQP4-IgG, and Multiple Sclerosis Optic Neuritis[1 ,15-17 ,20]
Characteristics
MOG-IgG-positive ON
AQP4-IgG-positive ON
Multiple sclerosis ON
Demographics
Bimodal: 30s and children
Female ~ Male
40s
Female
20s
Female
Pathophysiology
Preservation of astrocytes
Damage to astrocytes
Preservation of astrocytes
Clinical Course
Eye pain
Bilateral simultaneous
Very poor visual acuity at nadir
Rapid recovery
Recurrent ON
Good final visual outcome
Eye pain
Bilateral simultaneous
Very poor visual acuity at nadir
Rapid recovery rarely seen
Recurrent ON
Poor final visual outcome
Eye pain
Bilateral simultaneous less frequent
Fair visual acuity at nadir
Rapid recovery sometimes seen
Recurrent ON sometimes seen
Good final visual outcome
Imaging and Work Up
Involves anterior optic pathway with >50% optic nerve length enhancement
Perineural enhancement
CSF IgG oligoclonal bands rarely seen
May see increased CNS neutrophils
Involves posterior optic pathway (predilection for optic tract and optic chiasm)
CSF IgG oligoclonal bands rarely seen
Periventricular white matter lesions
Elevated oligoclonal IgG bands
Radiologic and Laboratory Work-up:
The development of live transfected cell-based assays has led to the ability to detect MOG antibodies, a highly specific biomarker for MOG-IgG ON. The sensitivity and specificity for MOG antibodies in distinguishing MOG-IgG ON from AQP4-Ab ON and MS is approximately 24% and 100%, respectively.[21] Testing for MOG-IgG should be based on serum using either flow cytometry or immunofluorescence. Enzyme-linked immunosorbent assays and immunoblotting are not recommended due to lack of specificity.[22]
On orbital MRI with fat suppression and gadolinium enhancement, there is typically predilection for the anterior optic pathway in patients with MOG-IgG ON. Typically, MOG-IgG ON involves longer segments of the optic nerve than in multiple sclerosis ON. Approximately 80% of patients with MOG-IgG positive ON have enhancement of more than 50% of the length of the optic nerve.[11] In addition, enhancement of the optic nerve sheath (i.e. perineural) is generally seen in 50% of MOG-IgG ON. In contrast, AQP4-IgG positive ON generally has a predilection for the posterior optic pathway. Similarly, long segments of the optic nerve may be involved, but it can also involve the optic tract and/or optic chiasm. [1 ,11] This is a key distinguishing feature between these entities, and therefore, illustrates the importance of obtaining orbital MRI in the workup of all patients presenting with optic neuritis.
MRI brain is abnormal in 45-77% of patients with MOG-IgG ON. Typically bilateral lesions are seen at onset (about 50% of cases). Periventricular white matter lesions (Dawson’s fingers) and U- or S-shape lesions seen in MS are uncommon. Other characteristic features may include: diffuse gray matter involvement, diffuse brainstem involvement, and multifocal white matter lesions. In children, cerebellar peduncle lesions may also been seen.[23]
CSF findings for MOG-IgG ON are less specific but may show pleocytosis and elevated protein..[20] Glial fibrillary acidic protein (GFAP) levels are generally elevated in the CSF of AQP4-IgG seropositive patients but not observed in the CSF of MOG-IgG seropositive patients.[24] Importantly, studies show that CSF IgG oligoclonal bands, a hallmark in multiple sclerosis, are rarely seen in MOG-IgG or AQP4-IgG seropositive patients.[1]
Table 2: Signs of possible MOG-IgG or AQP4-IgG Optic Neuritis
Consider testing serum for MOG-IgG and/or AQP4-IgG in patients with ON and the following:
Severely impaired visual acuity at onset (<20/200)
Bilateral or recurrent optic neuritis (more than twice per year)
Prominent disc edema (more common in MOG-IgG ON)
Poor visual recovery (visual acuity fails to improve beyond 20/100)
Longitudinally extensive optic nerve (>50% of length) MRI enhancement
Optic nerve sheath (i.e. perineural) enhancement
CSF oligoclonal bands not observed
MRI brain do not show characteristic multiple sclerosis findings
Treatment/Management/Guidelines:
In contrast, several studies have suggested that early plasma exchange results in better outcomes for those with AQP4-IgG positive ON.[25] In addition, long-term immunotherapy is usually recommended for patients with AQP4-IgG positive ON. Rituximab has been used as a first-line chronic immunomodulation for these patients in combination with IV immunoglobulin treatment, however azathioprine or mycophenolate mofetil may also be considered.[1] Thus, it is crucial to identify these biomarkers early in the disease course since it can significantly alter the choice of therapy, and ultimately, the overall management plan.
EPIDEMIOLOGY
Bimodal: 30s and children
No sex predilection
More common in Caucasian
Main Clinical Featues
Significant eye pain
Bilateral simultaneous optic neuritis
Very poor visual acuity at presentation
High relapse rate
Good visual recovery
Laboratory & Imaging Findings
Positive serum MOG-IgG using a transfected cell-based assay
CSF analysis:
Pleocytosis (~50%)
Oligoclonal bands very rare
Optic nerve MRI:
Enhancement of >50% of optic nerve length
Orbital portion involved
Perineural enhancement
Chiasm rarely involved
Optic tract rarely involved
Brain MRI:
Deep gray matter involvement
Diffuse brainstem involvement
Multifocal white matter lesions
Dawson’s fingers uncommon
TREATMENT/MANAGEMENT
Acute:
IV steroids followed by PO prednisone taper
Consider plasma exchange or IVIG if vision loss is severe or failure of recovery after initiating high dose steroids[1]
Chronic:
Consider immunotherapy (rituximab, azathioprine, mycophenolate mofetil) for single attack with significant disability, recurrent demyelinating attacks, or if MOG-IgG still positive at 6 months
References
1. Chen JJ, Pittock SJ, Flanagan EP, Lennon VA, Bhatti MT. Optic neuritis in the era of biomarkers. Surv Ophthalmol 2020;65(1):12-17. https://PubMed.gov/31425702. DOI: 10.1016/j.survophthal.2019.08.001
3. Jitprapaikulsan J, Chen JJ, Flanagan EP, Tobin WO, Fryer JP, Weinshenker BG, McKeon A, Lennon VA, Leavitt JA, Tillema JM, Lucchinetti C, Keegan BM, Kantarci O, Khanna C, Jenkins SM, Spears GM, Sagan J, Pittock SJ. Aquaporin-4 and Myelin Oligodendrocyte Glycoprotein Autoantibody Status Predict Outcome of Recurrent Optic Neuritis. Ophthalmology 2018;125(10):1628-1637. https://PubMed.gov/29716788. DOI: 10.1016/j.ophtha.2018.03.041
4. Ducloyer JB, Caignard A, Aidaoui R, Ollivier Y, Plubeau G, Santos-Moskalyk S, Porphyre L, Le Jeune C, Bihl L, Alamine S, Marignier R, Bourcier R, Ducloyer M, Weber M, Le Meur G, Wiertlewski S, Lebranchu P. MOG-Ab prevalence in optic neuritis and clinical predictive factors for diagnosis. Br J Ophthalmol 2019;10.1136/bjophthalmol-2019-314845. https://PubMed.gov/31582363. DOI: 10.1136/bjophthalmol-2019-314845
5. Jurynczyk M, Messina S, Woodhall MR, Raza N, Everett R, Roca-Fernandez A, Tackley G, Hamid S, Sheard A, Reynolds G, Chandratre S, Hemingway C, Jacob A, Vincent A, Leite MI, Waters P, Palace J. Clinical presentation and prognosis in MOG-antibody disease: a UK study. Brain 2017;140(12):3128-3138. https://PubMed.gov/29136091. DOI: 10.1093/brain/awx276
6. Fernandez-Carbonell C, Vargas-Lowy D, Musallam A, Healy B, McLaughlin K, Wucherpfennig KW, Chitnis T. Clinical and MRI phenotype of children with MOG antibodies. Mult Scler 2016;22(2):174-184. https://PubMed.gov/26041801. DOI: 10.1177/1352458515587751
7. Hacohen Y, Wong YY, Lechner C, Jurynczyk M, Wright S, Konuskan B, Kalser J, Poulat AL, Maurey H, Ganelin-Cohen E, Wassmer E, Hemingway C, Forsyth R, Hennes EM, Leite MI, Ciccarelli O, Anlar B, Hintzen R, Marignier R, Palace J, Baumann M, Rostasy K, Neuteboom R, Deiva K, Lim M. Disease Course and Treatment Responses in Children With Relapsing Myelin Oligodendrocyte Glycoprotein Antibody-Associated Disease. JAMA Neurol 2018;75(4):478-487. https://PubMed.gov/29305608. DOI: 10.1001/jamaneurol.2017.4601
8. Rostasy K, Mader S, Schanda K, Huppke P, Gartner J, Kraus V, Karenfort M, Tibussek D, Blaschek A, Bajer-Kornek B, Leitz S, Schimmel M, Di Pauli F, Berger T, Reindl M. Anti-myelin oligodendrocyte glycoprotein antibodies in pediatric patients with optic neuritis. Arch Neurol 2012;69(6):752-756. https://PubMed.gov/22371853. DOI: 10.1001/archneurol.2011.2956
9. Chen Q, Zhao G, Huang Y, Li Z, Sun X, Lu P, San Y, Wang M, Tian G. Clinical Characteristics of Pediatric Optic Neuritis With Myelin Oligodendrocyte Glycoprotein Seropositive: A Cohort Study. Pediatr Neurol 2018;83:42-49. https://PubMed.gov/29778487. DOI: 10.1016/j.pediatrneurol.2018.03.003
10. Song H, Zhou H, Yang M, Tan S, Wang J, Xu Q, Liu H, Wei S. Clinical characteristics and prognosis of myelin oligodendrocyte glycoprotein antibody-seropositive paediatric optic neuritis in China. Br J Ophthalmol 2019;103(6):831-836. https://PubMed.gov/30049802. DOI: 10.1136/bjophthalmol-2018-312399
11. Chen JJ, Flanagan EP, Jitprapaikulsan J, Lopez-Chiriboga ASS, Fryer JP, Leavitt JA, Weinshenker BG, McKeon A, Tillema JM, Lennon VA, Tobin WO, Keegan BM, Lucchinetti CF, Kantarci OH, McClelland CM, Lee MS, Bennett JL, Pelak VS, Chen Y, VanStavern G, Adesina OO, Eggenberger ER, Acierno MD, Wingerchuk DM, Brazis PW, Sagen J, Pittock SJ. Myelin Oligodendrocyte Glycoprotein Antibody-Positive Optic Neuritis: Clinical Characteristics, Radiologic Clues, and Outcome. Am J Ophthalmol 2018;195:8-15. https://PubMed.gov/30055153. DOI: 10.1016/j.ajo.2018.07.020
12. Johns TG, Bernard CC. The structure and function of myelin oligodendrocyte glycoprotein. J Neurochem 1999;72(1):1-9. https://PubMed.gov/9886048. DOI: 10.1046/j.1471-4159.1999.0720001.x
13. Weissert R, Kuhle J, de Graaf KL, Wienhold W, Herrmann MM, Müller C, Forsthuber TG, Wiesmüller KH, Melms A. High immunogenicity of intracellular myelin oligodendrocyte glycoprotein epitopes. J Immunol 2002;169(1):548-556. https://PubMed.gov/12077287. DOI: 10.4049/jimmunol.169.1.548
14. Lennon VA, Kryzer TJ, Pittock SJ, Verkman AS, Hinson SR. IgG marker of optic-spinal multiple sclerosis binds to the aquaporin-4 water channel. J Exp Med 2005;202(4):473-477. https://PubMed.gov/16087714. DOI: 10.1084/jem.20050304
15. Ikeda K, Kiyota N, Kuroda H, Sato DK, Nishiyama S, Takahashi T, Misu T, Nakashima I, Fujihara K, Aoki M. Severe demyelination but no astrocytopathy in clinically definite neuromyelitis optica with anti-myelin-oligodendrocyte glycoprotein antibody. Mult Scler 2015;21(5):656-659. https://PubMed.gov/25257613. DOI: 10.1177/1352458514551455
16. Spadaro M, Gerdes LA, Mayer MC, Ertl-Wagner B, Laurent S, Krumbholz M, Breithaupt C, Högen T, Straube A, Giese A, Hohlfeld R, Lassmann H, Meinl E, Kümpfel T. Histopathology and clinical course of MOG-antibody-associated encephalomyelitis. Ann Clin Transl Neurol 2015;2(3):295-301. https://PubMed.gov/25815356. DOI: 10.1002/acn3.164
17. Dos Passos GR, Oliveira LM, da Costa BK, Apostolos-Pereira SL, Callegaro D, Fujihara K, Sato DK. MOG-IgG-Associated Optic Neuritis, Encephalitis, and Myelitis: Lessons Learned From Neuromyelitis Optica Spectrum Disorder. Front Neurol 2018;9:217. https://PubMed.gov/29670575. DOI: 10.3389/fneur.2018.00217
19. Pache F, Zimmermann H, Mikolajczak J, Schumacher S, Lacheta A, Oertel FC, Bellmann-Strobl J, Jarius S, Wildemann B, Reindl M, Waldman A, Soelberg K, Asgari N, Ringelstein M, Aktas O, Gross N, Buttmann M, Ach T, Ruprecht K, Paul F, Brandt AU. MOG-IgG in NMO and related disorders: a multicenter study of 50 patients. Part 4: Afferent visual system damage after optic neuritis in MOG-IgG-seropositive versus AQP4-IgG-seropositive patients. J Neuroinflammation 2016;13(1):282. https://PubMed.gov/27802824. DOI: 10.1186/s12974-016-0720-6
20. Kothur K, Wienholt L, Tantsis EM, Earl J, Bandodkar S, Prelog K, Tea F, Ramanathan S, Brilot F, Dale RC. B Cell, Th17, and Neutrophil Related Cerebrospinal Fluid Cytokine/Chemokines Are Elevated in MOG Antibody Associated Demyelination. PLoS One 2016;11(2):e0149411. https://PubMed.gov/26919719. DOI: 10.1371/journal.pone.0149411
21. Waters P, Woodhall M, O'Connor KC, Reindl M, Lang B, Sato DK, Juryńczyk M, Tackley G, Rocha J, Takahashi T, Misu T, Nakashima I, Palace J, Fujihara K, Leite MI, Vincent A. MOG cell-based assay detects non-MS patients with inflammatory neurologic disease. Neurol Neuroimmunol Neuroinflamm 2015;2(3):e89. https://PubMed.gov/25821844. DOI: 10.1212/nxi.0000000000000089
22. Tajfirouz DA, Bhatti MT, Chen JJ. Clinical Characteristics and Treatment of MOG-IgG-Associated Optic Neuritis. Curr Neurol Neurosci Rep 2019;19(12):100. https://PubMed.gov/31773369. DOI: 10.1007/s11910-019-1014-z
23. Wynford-Thomas R, Jacob A, Tomassini V. Neurological update: MOG antibody disease. J Neurol 2019;266(5):1280-1286. https://PubMed.gov/30569382. DOI: 10.1007/s00415-018-9122-2
24. Kaneko K, Sato DK, Nakashima I, Ogawa R, Akaishi T, Takai Y, Nishiyama S, Takahashi T, Misu T, Kuroda H, Tanaka S, Nomura K, Hashimoto Y, Callegaro D, Steinman L, Fujihara K, Aoki M. CSF cytokine profile in MOG-IgG+ neurological disease is similar to AQP4-IgG+ NMOSD but distinct from MS: a cross-sectional study and potential therapeutic implications. J Neurol Neurosurg Psychiatry 2018;89(9):927-936. https://PubMed.gov/29875186. DOI: 10.1136/jnnp-2018-317969
25. Abboud H, Petrak A, Mealy M, Sasidharan S, Siddique L, Levy M. Treatment of acute relapses in neuromyelitis optica: Steroids alone versus steroids plus plasma exchange. Mult Scler 2016;22(2):185-192. https://PubMed.gov/25921047. DOI: 10.1177/1352458515581438
Suggested citation format:
Diel RJ, Shah T, Kardon RH, Wall M. Myelin Oligodendrocyte Glycoprotein (MOG)-IgG Associated Optic Neuritis EyeRounds.org. Posted September 17, 2020. Available from 301-MOG-optic-neuritis.htm.
University of Iowa
Roy J. and Lucille A. Carver College of Medicine
Department of Ophthalmology and Visual Sciences
200 Hawkins Drive
Iowa City, IA 52242
University of Iowa
Roy J. and Lucille A. Carver College of Medicine
Department of Ophthalmology and Visual Sciences
200 Hawkins Drive
Iowa City, IA 52242




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}