Chief Complaint: “I was told that I have a retinal scar in my right eye.”
History of Present Illness (HPI)
The patient is a 58-year-old African American woman with an ocular history of mild primary open angle glaucoma and age-related cataracts in both eyes who presents for the evaluation of a newly-noted retinal scar in her right eye. The scar was found incidentally during a routine exam by her optometrist. She is asymptomatic with good vision in both eyes and is unaware of any changes in her vision. She also denies episodes of flashes of light, floaters, visual field shadowing, pain with eye movements, light sensitivity, or night blindness.
Past Ocular History
Past Medical History
Medications
Allergies
Family History
Social History
Review of Systems
OCULAR EXAMINATION
| OD | OS | |
|---|---|---|
| Lids/Lashes | Normal | Normal |
| Conunctiva/Sclera | Limbal complexion associated melanosis | Limbal complexion associated melanosis |
| Cornea | Clear | Clear |
| Anterior Chamber | Deep and quiet | Deep and quiet |
| Iris | Normal architecture | Normal architecture |
| Lens | Trace nuclear sclerosis | Trace nuclear sclerosis |
| OD | OS | |
|---|---|---|
| Vitreous | No anterior vitreous cell | No anterior vitreous cell |
| Disc | No edema, pallor, or hemorrhages | No edema, pallor, or hemorrhages |
| Cup-to-disc ratio | 0.45 | 0.45 |
| Macula | Blunted foveal reflex | Blunted foveal reflex |
| Vessels | Normal | Normal |
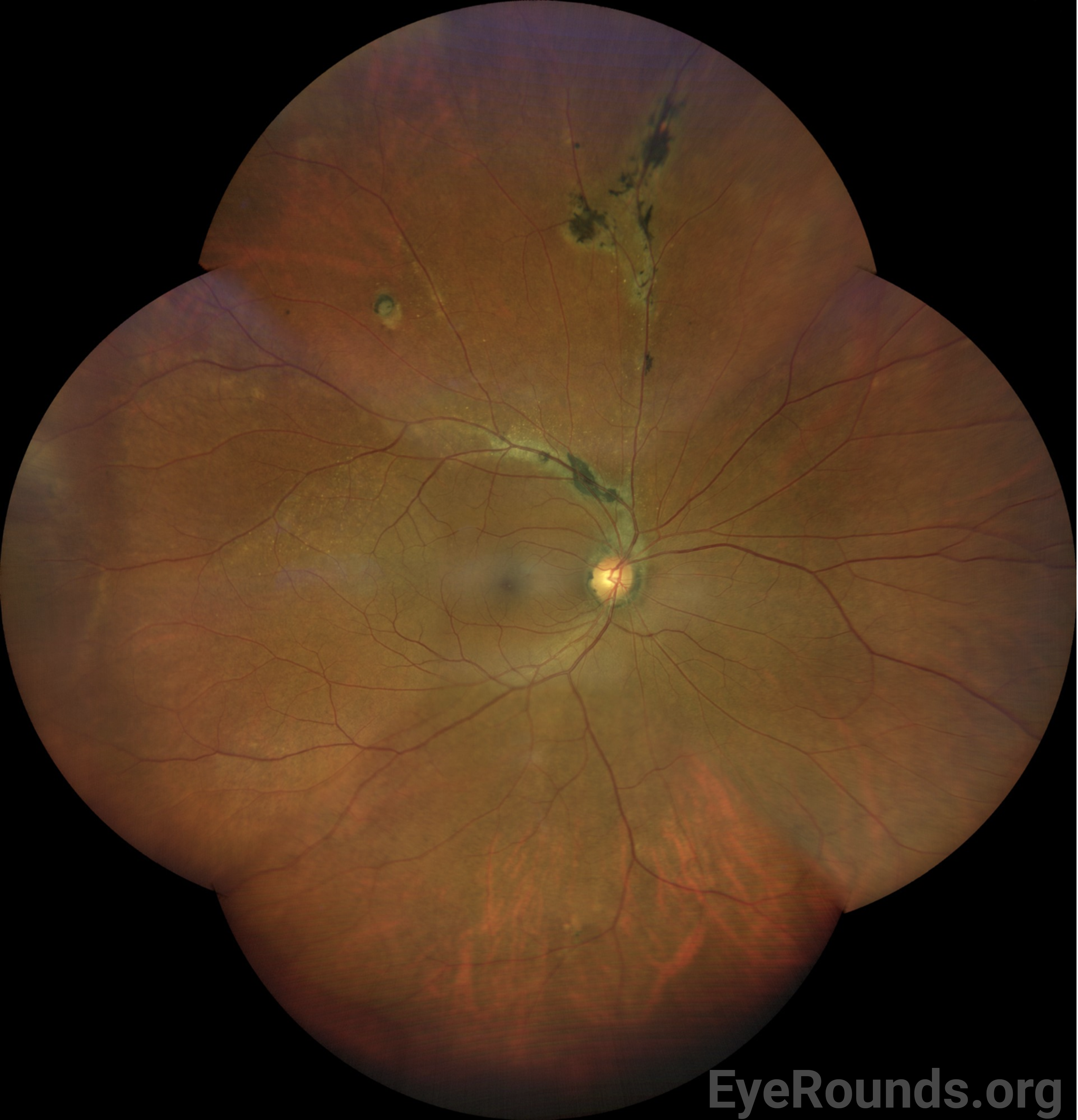
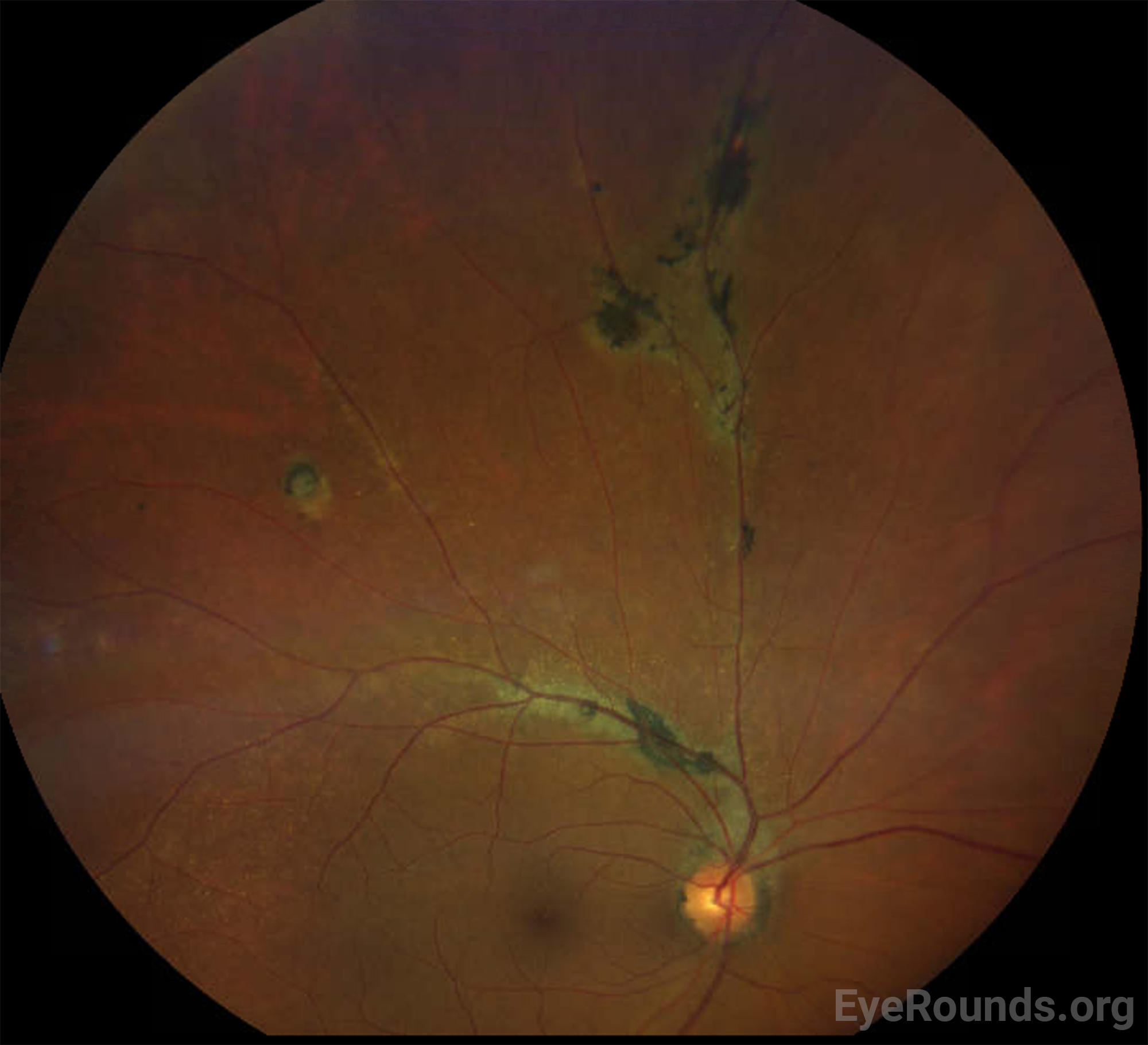
| Periphery | Multifocal hyperpigmented chorioretinal scars and atrophy with perivenous predilection along branches of the superior arcade. Circular chorioretinal scar and atrophy along the supertemporal mid-periphery. | Normal |
Differential Diagnosis
CLINICAL COURSE
The patient was asymptomatic on presentation and was at her baseline visual acuity. Given the non-central location of the chorioretinal scarring and the lack of associated signs to suggest current or prior inflammation or other concomitant ocular disease, the patient was diagnosed with pigmented paravenous retinochoroidal atrophy, and reassurance was provided for annual monitoring for any progression.
DIAGNOSIS: Pigmented paravenous retinochoroidal atrophy
DISCUSSION
Etiology
Pigmented paravenous retinochoroidal atrophy (PPRCA) is a rare condition characterized by paravenous pigment aggregations along the retinal veins associated with adjacent chorioretinal atrophy. Most cases of PPRCA are considered isolated or sporadic, with no known underlying inherited or inflammatory cause identified [1-3]. However, similar perivenular pigmentation can be seen in those with inherited retinal diseases [4-6], including CRB1-associated retinitis pigmentosa (RP) or Leber congenital amaurosis (LCA) [7,8], or in the context of abnormal embryonic ocular development such as macular coloboma [8,9]. Paravenous pigmentation can also be seen in inflammatory and infectious diseases, including tuberculosis, syphilis, sarcoidosis, Vogt-Koyanagi-Harada disease, Behcet’s disease, measles, and rubella [10-13].
Pathophysiology and Natural History
PPRCA is highlighted by pigment aggregation and clumping along the retinal veins leading to zonal retinochoroidal atrophy, with resultant atrophic and eventual visual changes [11]. The exact pathophysiology of these changes remains poorly understood, but dysfunction of the retinal pigment epithelium (RPE) is thought to play a role in photoreceptor degeneration as well as pigmentary abnormalities [9,11].
Signs/Symptoms
PPRCA is often an incidental finding without associated symptoms. Some patients experience mild blurred vision, but severely reduced vision can be seen when the macula is involved. Funduscopic examination reveals coarse pigmentary clumps within the retina, fine pigmentary changes along the retinal veins, and bone spicule-like pigmentation, along with retinochoroidal atrophy, usually away from the optic nerve head. Typically, the pigmentation extends alongside the posterior pole of the retina and into the periphery. PPRCA is often bilateral but asymmetric [14], but unilateral presentations have also been reported [15-17]. While macular involvement is rare, some have reported associated pigmentary macular degeneration with RPE atrophy, lamellar macular hole formation, and cystoid macular edema [18].
Diagnosis
PPRCA is diagnosed by clinical examination and the exclusion of other potential underlying causes through a variety of different diagnostic tests that can be considered in workup, including fundus autofluorescence (FAF) [19], fluorescein angiography (FA) [19,20], indocyanine green angiography (ICG) [21], electroretinogram (ERG) [11], and optical coherence tomography (OCT) [22,23], and OCT angiography (OCTA)[24,25].
FAF can be helpful to demonstrate the extent of PPRCA, with areas of hypo-autofluorescence corresponding to areas of RPE atrophy, and hyper-autofluorescence corresponding to pigment clumping and areas of dysfunctional RPE. When feasible to obtain (as PPRCA is often peripheral), OCT may show areas of outer retinal and RPE loss corresponding to the pigmented lesions on clinical examination. FA and ICG may show hyperfluorescence from window defects in areas of retinochoroidal atrophy and can be considered in cases with suspicion for an underlying inflammatory condition (e.g., to evaluate for subclinical chorioretinal lesions or assess for vascular leakage). ERG may be considered in cases where an underlying photoreceptor degeneration may be present (e.g., retinitis pigmentosa). OCTA has been used to identify the extent of retinal and choroidal vascular injury related to PPRCA, with suggestion of vascular compromise to choriocapillaris and the deep capillary plexus playing a role [24,25].
Treatment
There is no current treatment for PPRCA.
EPIDEMIOLOGY
|
ETIOLOGY
|
SYMPTOMS/SIGNSSymptoms
Signs
|
Diagnosis
|
Gehrke E, Strampe MR, Drack AV. Diagnosis and management of Juvenile X-linked Retinoschisis (JXLR). EyeRounds.org. Posted December 5, 2023; Available from https://EyeRounds.org/cases/354-Morning-Glory-Disc-Anomaly.htm

Ophthalmic Atlas Images by EyeRounds.org, The University of Iowa are licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License.

Address
University of IowaLegal
Related Links