
INITIAL PRESENTATION
Chief Complaint: Eyes “shaking” shortly after birth
History of Present Illness
The patient is a 4-month-old female who was referred to the pediatric ophthalmology clinic due to concern for oculocutaneous albinism (OCA). She was born at 39 weeks gestation via Cesarian section, and her growth and development have been normal to this point. Her parents have noted that her eyes “shake” since shortly after birth. However, they deny crossing or drifting of the eyes. They report that she makes eye contact intermittently and likes to look at faces and her mobile hanging from the ceiling. Per their observations, she seems to be quite sensitive to bright lights. She is blonde but looks “just like her brother at that age.” Neither her brother nor other family members have significant ocular history.
Past Ocular History
Past Medical History
Medications
Allergies
Family History
Social History
Review of Systems
OCULAR EXAMINATION
DIFFERENTIAL DIAGNOSIS:
Disorders associated with early-onset nystagmus
Disorders associated with hypopigmentation of the skin and fundus, and foveal hypoplasia
CLINICAL COURSE
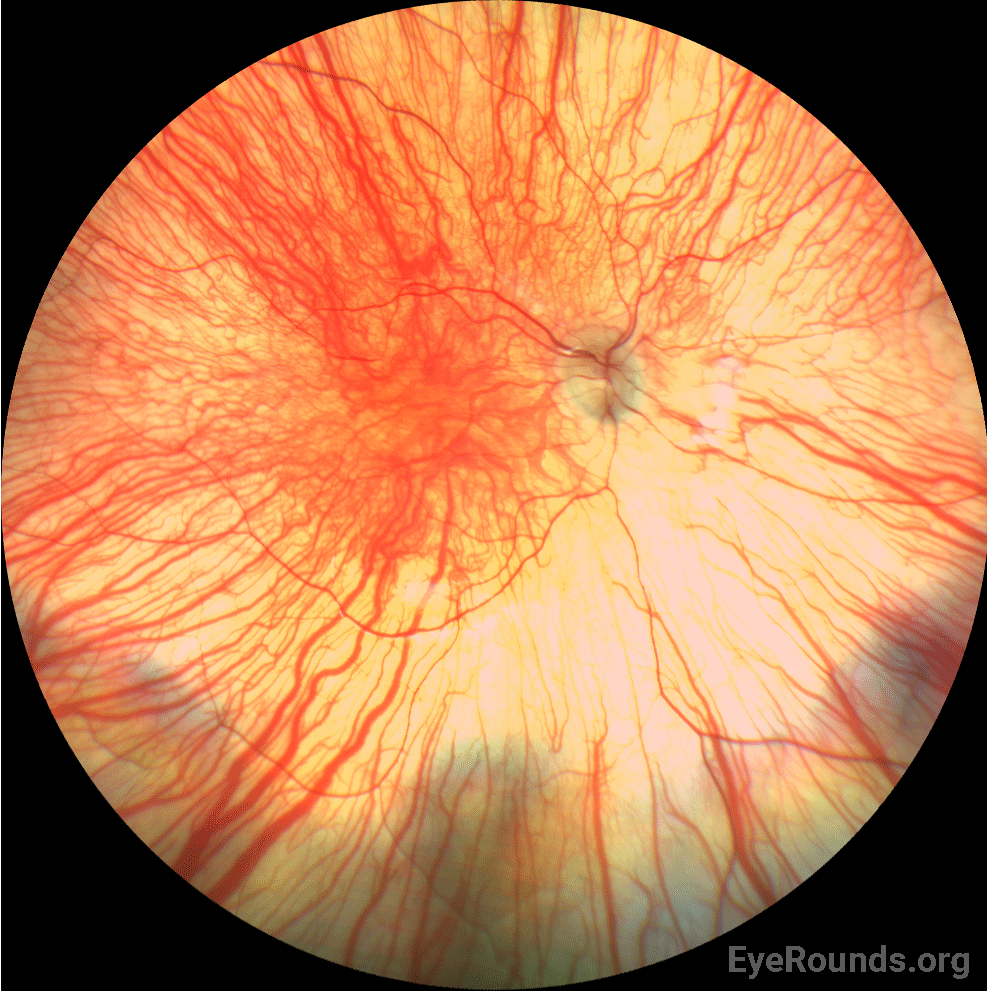
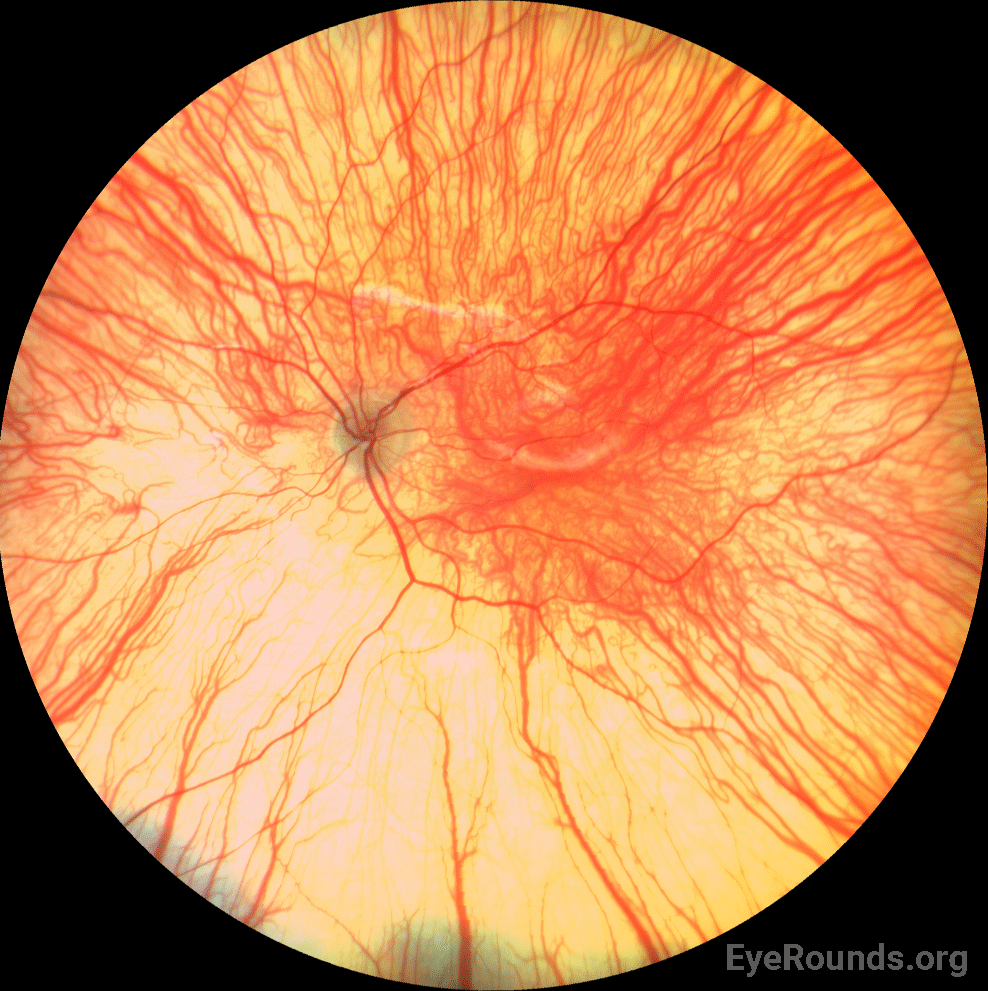
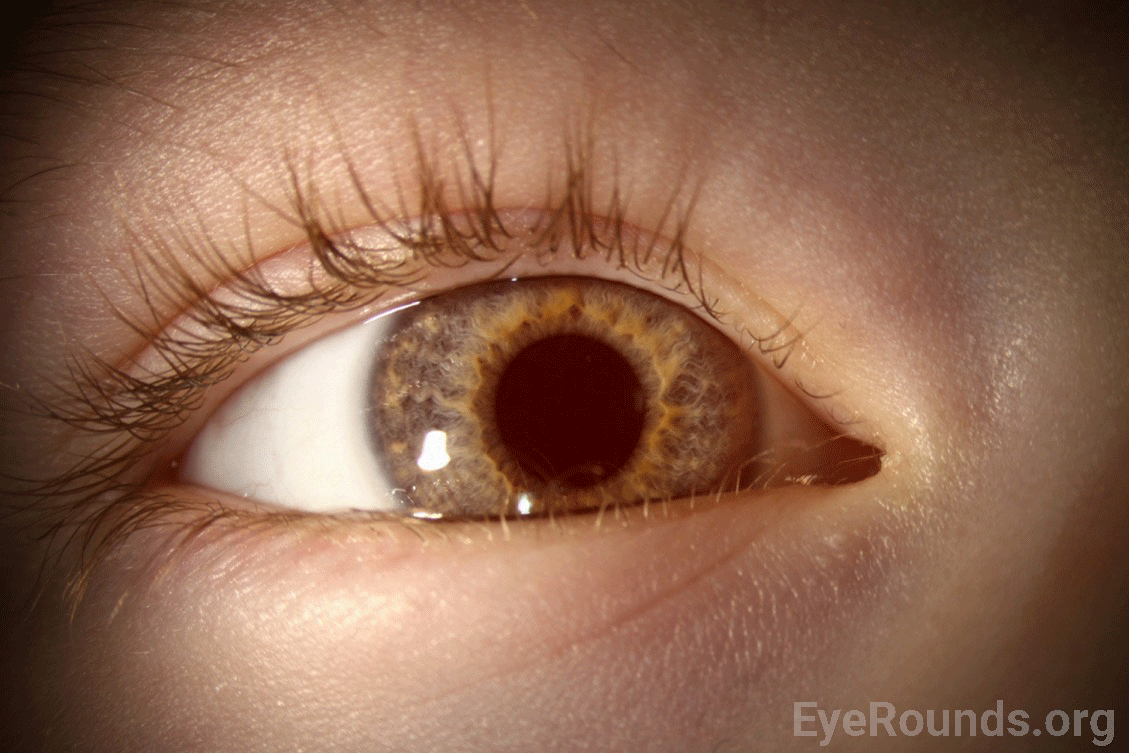
Given the patient’s complete iris TID’s, nystagmus, and poor foveal reflex, oculocutaneous albinism was thought to be the most likely diagnosis. An albinism panel was obtained which showed no mutations found in any OCA gene. 3 months later, she was seen in clinic for a repeat vision check and retinoscopy given her relatively high myopia. By the time she returned for this visit, she had been seen at the Mayo Clinic and underwent platelet electron microscopy. This was significant for lack of platelet-dense granules, suggestive of Hermansky-Pudlak Syndrome (HPS). She was referred to hematology clinic for further evaluation for potential HPS. Upon further questioning, her parents endorsed a history of easy bruising. Genetic testing for HPS was obtained, which revealed compound heterozygous pathogenic variants in HPS5.
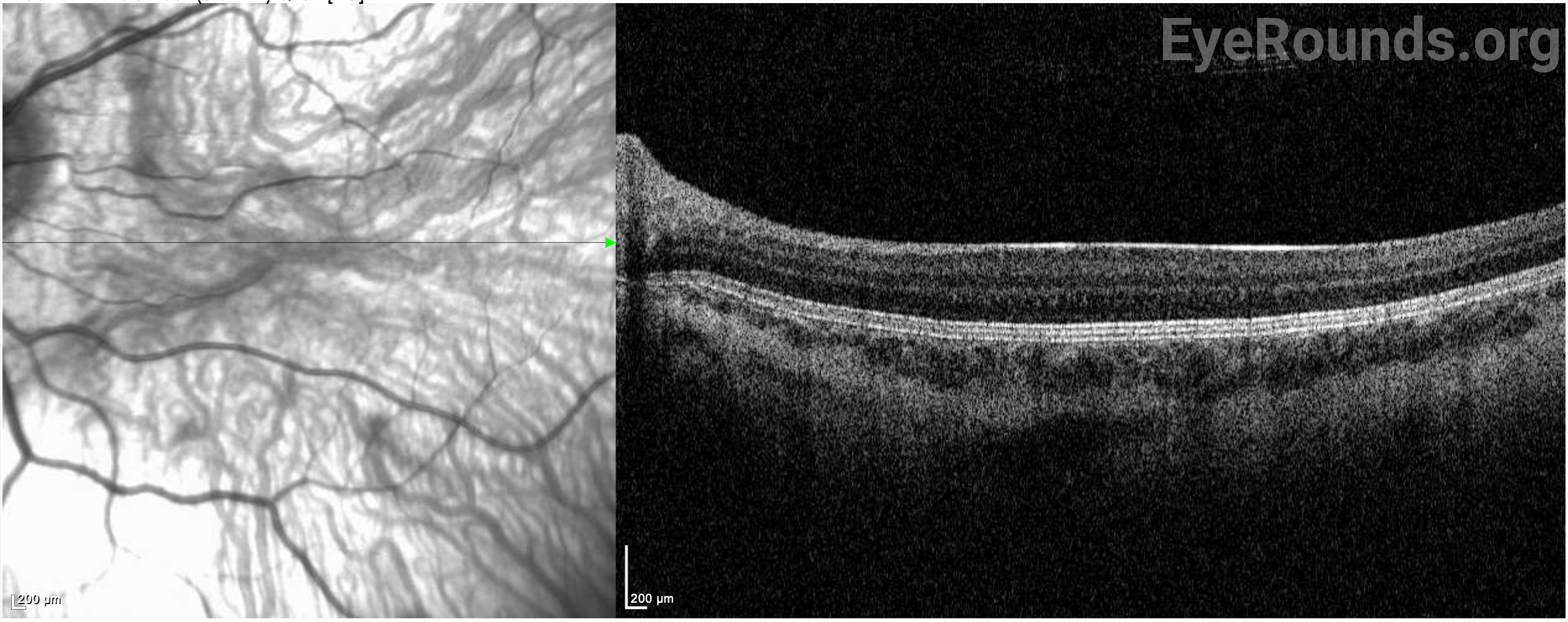
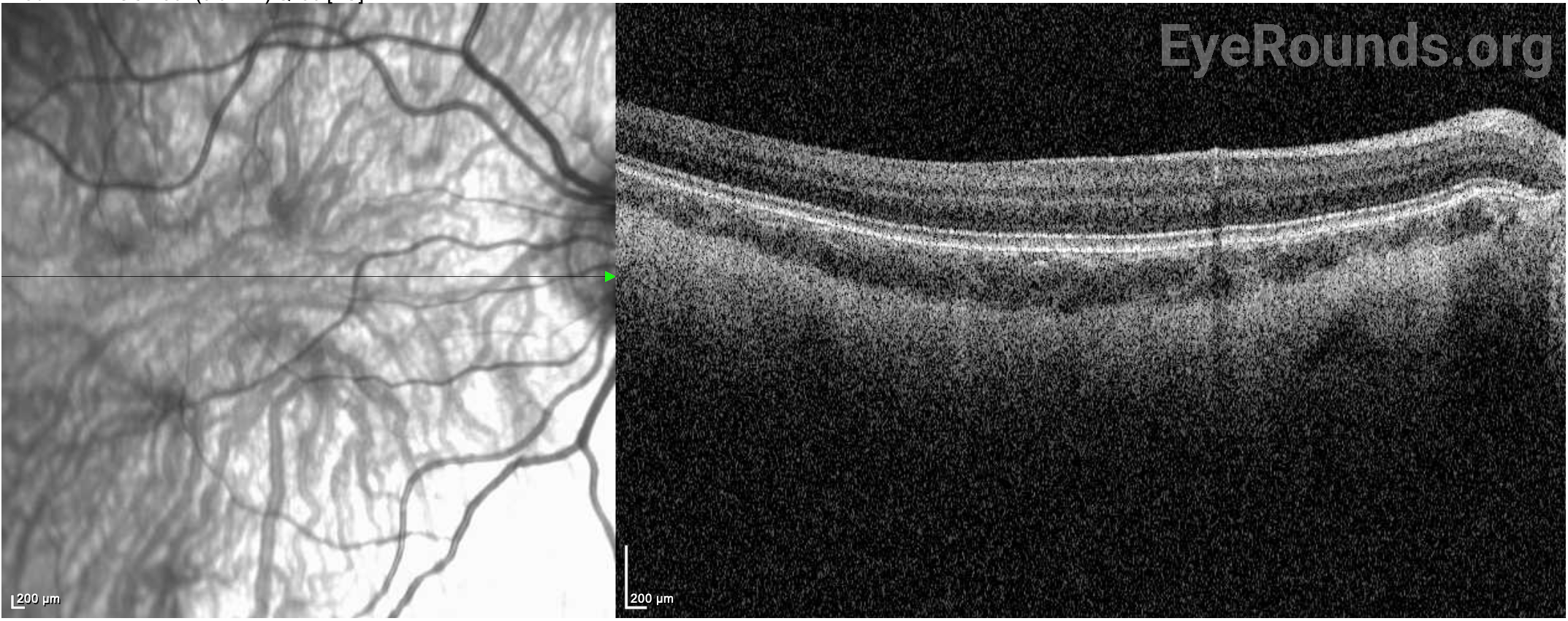
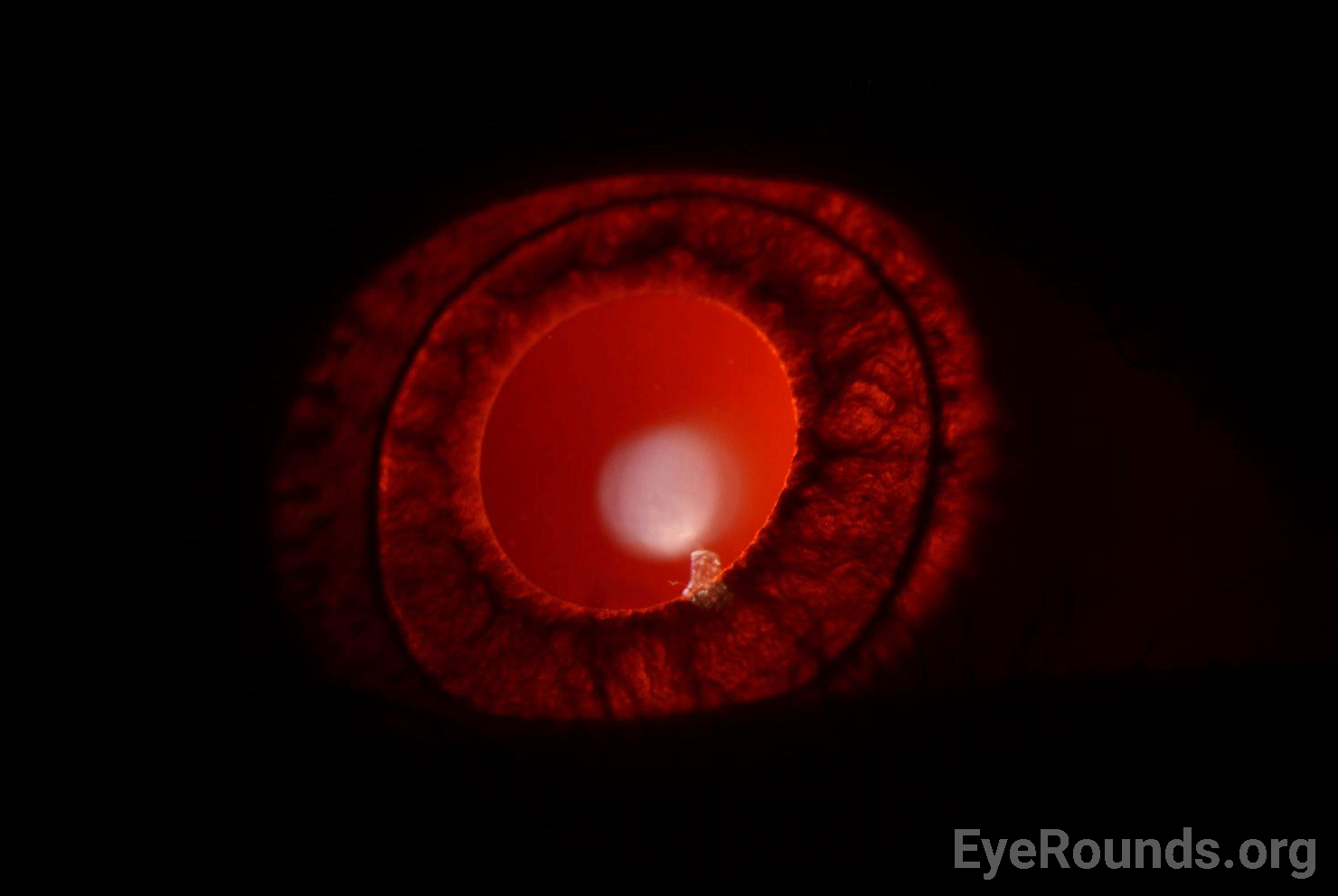
She continued to follow up regularly with the pediatric ophthalmology clinic for treatment of her myopia and regular astigmatism. She was treated with glasses for her myopia and transition lenses for photophobia. She was noted to have continued nystagmus and a compensatory chin-down abnormal head position. She was offered Kestenbaum surgery for treatment of her abnormal head position, which the family declined due to concern for bleeding and desire to let her develop further. Her myopia improved with glasses, but her vision continued to be limited by severe foveal hypoplasia, confirmed on optical coherence tomography (OCT).
She also was followed by the comprehensive bleeding disorders clinic and was treated with iron supplementation. Her bleeding symptoms remained mild, with occasional bruising and epistaxis not requiring treatment. Additionally, she received several months of developmental and vision therapy, including visits with the low-vision specialist. She received assistance with a monocular telescope, lighted hand magnifier, and an iPad for school. Additionally, she received binoculars for riding in the car and trips to the zoo.
On her most recent visit to pediatric ophthalmology (at age 8), her best corrected visual acuity was 20/150 in both eyes. She wore her glasses daily with a prescription of -1.75 + 3.75 x 087 OD and -1.00 + 4.50 x 091 OS. Her parents felt like her vision had improved, and her nystagmus and compensatory head position were both less noticeable. A plan was made for her to continue yearly follow-ups, as well as skin and eye protection from sunlight.
DIAGNOSIS: Hermansky-Pudlak Syndrome, Type 5
DISCUSSION
Etiology/Epidemiology
Hermansky-Pudlak Syndrome (HPS) is a genetic disorder that is inherited in an autosomal recessive manner (2). To date, eleven subtypes of HPS have been identified (3). Each subtype corresponds to one of eleven identified pathogenic variants in the HPS gene (4). Common features of the syndrome across all subtypes include clinical features of OCA with various degrees of hypopigmentation of skin and hair, congenital nystagmus, hypopigmentation of the uveal tract (iris transillumination and decreased fundus pigmentation), and foveal hypoplasia as well as a mild to severe bleeding diathesis from platelet dysfunction. Several other systemic features may be present such as pulmonary fibrosis, granulomatous colitis, renal impairment, and cardiomyopathy; however, these vary between subtypes and are not always present (2).
While the exact incidence of HPS is unknown, over 1200 patients are registered with HPS Network, Inc., a non-profit organization supporting patients with HPS (2). HPS has been described across multiple ethnic and geographic groups but, due to founder mutations, has an especially high incidence in Puerto Rican and Ashkenazi Jewish populations (5). In northwest Puerto Rico, 1 in 1800 people have HPS type 1 (HPS-1), and 1 in 21 people is a carrier for the founder mutation (a 16 base-pair duplication in exon 15 of the HPS1 gene). In central Puerto Rico, 1 in 4000 people have HPS-3 (2).
Pathophysiology
Proteins encoded by the HPS gene assemble into four different complexes: biogenesis of lysosome-related organelle complex (BLOC) 1-3 and adaptor protein (AP)-3 (6). This leads to deficits in intracellular protein trafficking and the biogenesis of lysosomes and lysosome-related organelles (LROs) (5). LROs are organelles derived from endosomes that have different structures and functions based on cell type (6). These include melanosomes, platelet-dense granules, and Weibel-Palade bodies in vascular endothelial cells (3,4). AP-3 has a role in transporting tyrosine into melanosomes for melanin synthesis, while dense granules contain serotonin, calcium, and other molecules necessary for coagulation and platelet adhesion (3). Together, disturbances in these processes lead to hallmark deficits in manufacturing and storing melanin and platelet aggregation (4).
In the eye, deficits in melanin production and storage lead to hypopigmentation of the iris and the retinal pigment epithelium (RPE), resulting in ocular manifestations such as reduced visual acuity and photophobia (7). Patients with OCA also have hypoplasia of the fovea. While the mechanism of foveal development is not entirely understood, several hypotheses have been proposed that lead to foveal hypoplasia. These include an imbalance of angiogenic and anti-angiogenic growth factors and dysfunctional signaling along several steps in the central pigmentation pathway (6).
The mechanisms behind other manifestations of HPS are also not completely understood. Lysosomal trafficking abnormalities in HPS can also result in cell signaling dysfunction such as in neurotransmission, cell proliferation, and immune responses (8). Patients with HPS also develop ceroid lipofuscin pigment deposition in multiple organs. It has been hypothesized that this is an inflammatory process that may contribute to pulmonary fibrosis and colitis (5). It has also been hypothesized that abnormalities in fibroblast and macrophage function in HPS contribute to pulmonary fibrosis (2).
The mechanism of immunodeficiency in HPS-2 is also incompletely understood, though studies have shown impaired cytokine and chemokine release by antigen-presenting cells. It has also been proposed that HPS affects the lytic granules of cytotoxic T cells leading to dysfunction of these cells and natural killer (NK) cells (4).
Signs/Symptoms
All subtypes of HPS have shared features of OCA and bleeding diathesis. There is a significant phenotypic variation among the patients. Patients present with various degrees hypopigmentation of the hair and skin, which is often apparent at birth (2). Lack of melanin in the skin can predispose patients to solar keratosis, photo-aging, sunburns, squamous cell carcinoma, basal cell carcinoma, and melanoma (4).
In addition to skin hypopigmentation, patients have various degrees of hypopigmentation of the iris and retina. Clinical exam shows iris transillumination defects, decreased visual acuity, congenital nystagmus, foveal hypoplasia, errors in color vision, and photophobia (7,9). Visual acuity is usually in the range of 20/60-20/400 (7). Patients also often have abnormal decussation of fibers in the optic chiasm (10). Amblyopia, astigmatism, cataract formation, and elevated intraocular pressure have been described in patients with HPS. Some of these findings, such as astigmatism, are common in patients with HPS while others, such as cataract formation, are very rare (11).
Bleeding diathesis is a common feature across all HPS subtypes. Platelet dysfunction in HPS often presents in newborn males with prolonged bleeding following circumcision. Children can also have excessive bruising with minimal trauma once they reach the age of ambulation (5). Other possible presentations include gingival bleeding, prolonged bleeding after surgery, heavy menstrual bleeding, or postpartum hemorrhage (4).
While it does not occur in all subtypes of HPS, pulmonary fibrosis is one of the most feared and serious complications of HPS. It occurs most often in patients with HPS-1 but has also been described in HPS subtypes 2 and 4 (2). Pulmonary fibrosis in HPS has a bimodal age of onset, presenting either in the fourth or sixth decade, depending on the patient’s genotype (4). These patients present with clinical features of pulmonary fibrosis such as non-productive cough, exertional dyspnea, and dry crackles. Later in the course of disease, patients can also have hypoxia with exertion and at rest (5). Pulmonary fibrosis associated with HPS typically leads to respiratory failure and death within 10 years of onset (4).
Granulomatous colitis can also manifest in some subtypes of HPS. This has been described in HPS subtypes 1, 4, and 6 and affects about 15 % of patients with HPS (2,4). HPS-associated colitis is similar in both pathology and presentation to Crohn’s Disease and presents with crampy abdominal pain, fever, weight loss, malabsorption, and watery or bloody diarrhea (4,5).
Other rarer complications of HPS have been described in the literature including immunodeficiency and neutropenia in patients with HPS-2 as well as renal impairment and cardiomyopathy (4,12).
Clinical features are variable among patients and may not be obvious, particularly in young children.
Testing/Laboratory work-up
HPS should be suspected in all patients with clinical features of OCA. The gold standard for HPS diagnosis is genetic testing. Genetic testing for HPS is available independently or included in albinism/hypopigmentation panels. As clinical findings alone cannot distinguish HPS from non-syndromic OCA, testing for HPS is recommended in all patients. Pre-and post-test genetic counseling should be provided for patients and families. Additionally, electron microscopy of platelets can be performed. The absence of dense bodies with normal platelet count is supportive of a diagnosis of HPS (2,5).
Imaging
It is recommended that patients with HPS subtypes 1, 2, and 4 receive a baseline high-resolution CT scan (HRCT) of the chest in adolescence as that is when symptoms of pulmonary fibrosis typically would first appear (5). Findings are similar to idiopathic pulmonary fibrosis (IPF) but typically present at a younger age. These findings include reticular opacities, thickened intralobular septa, honeycombing, and ground-glass opacities (2).
Treatment/Management/Guidelines
To manage skin manifestations in HPS, patients should avoid sunlight during peak daytime hours, wear sun-protective clothing, and apply SPF-30 or higher sunscreen whenever they are outside (5). Additionally, patients should receive regular skin checks due to their increased risk of squamous cell carcinoma, basal cell carcinoma, and melanoma (4).
Similar guidance for avoiding sun exposure is recommended to protect the eyes of patients with HPS. These patients should also wear dark glasses or transition lenses for photophobia (5,7). All patients with HPS, especially in childhood, require correction of refractive error and treatment of amblyopia and strabismus if present. Some patients may require contact lenses or eye muscle surgery for treatment of abnormal head position associated with nystagmus. Based on their degree of visual impairment, patients should receive low vision aids for school and daily living (7).
Additional management of HPS depends on each patient's specific clinical features. Patients may require pre- and intra-operative management of bleeding complications; for example, with pre-operative desmopressin and platelet transfusions (4,5). It is recommended that patients with HPS-associated pulmonary fibrosis be referred for lung transplant early (2). Baseline HRCT is recommended in late adolescence; however, lung biopsy is not recommended due to the elevated risk for bleeding in these patients (2,5). The management of HPS-associated granulomatous colitis is similar to the management of Crohn’s disease and includes anti-inflammatory medications and immunosuppressants (2). Investigational treatment modalities such as stem cell and genetic therapies appear promising for OCA and its syndromic forms such as HPS but need to be studied further (6,8)
EPIDEMIOLOGY OR ETIOLOGY
|
DIAGNOSIS
|
SIGNS AND SYMPTOMS
|
TREATMENT/MANAGEMENT
|
References
Kuziel JD, Strampe MR, Dumitrescu AV. Hermansky-Pudlak Syndrome. EyeRounds.org. Jan. 26, 2024. Available from https://eyerounds.org/cases/350-hermansky-pudlak-syndrome.htm

Ophthalmic Atlas Images by EyeRounds.org, The University of Iowa are licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License.

Address
University of IowaLegal
Related Links

{kind=link}
{kind=link}