INITIAL PRESENTATION
Chief Complaint: Lower extremity weakness, double vision, and urinary incontinence
History of Present Illness
A 40-year-old female, 2 months status post Roux-en-Y presented to an outside hospital for 3 weeks of lower extremity weakness, double vision, and urinary incontinence.
One month after undergoing a complicated gastric bypass, she developed lower extremity weakness, numbness, and paresthesia to the degree that she could no longer ambulate. Upper extremity strength was unaffected. Over the same period, she reported double vision that was binocular and horizontal, as well as one episode of bladder incontinence. Prior to developing these symptoms, she endorsed persistent nausea and vomiting post-surgery. A review of her chart was notable for a 20 kg weight loss over the 2 months following her Roux-en-Y.
Her initial workup at an outside hospital included a CT head and vitamin B12 labs, both were within normal limits. Out of suspicion for Wernicke’s Encephalopathy, she empirically received 500 mg IV thiamine at the outside hospital without improvement. She was transferred to UIHC for further workup and management.
Past Ocular History
Past Medical History
Medications
Allergies
Family History
Social History
Review of Systems
OCULAR EXAMINATION
| -1 | -1 | -1 |
|---|---|---|
| -1.5 | 0 | |
| 0 | 0 | 0 |
| -1 | -1 | -1 |
|---|---|---|
| 0 | -1 | |
| 0 | 0 | 0 |
Eye movements: Abnormal saccades with evidence of horizontal and vertical gaze palsies.
Nystagmus: Primary position up-beat nystagmus (conjugate, jerk, low amplitude, moderate frequency), with a combination of up-beat and gaze-evoked patterns in eccentric gazes.
CLINICAL COURSE
Upon admission, the neurological exam was notable for ataxia, ophthalmoplegia with bilateral abduction deficits, vertical nystagmus, and hyporeflexia in bilateral lower extremities. Neurology was primarily concerned about nutritional deficiency causing WE, especially given recent bariatric surgery, as well as Guillan-Barre, specifically the Miller Fisher variant.
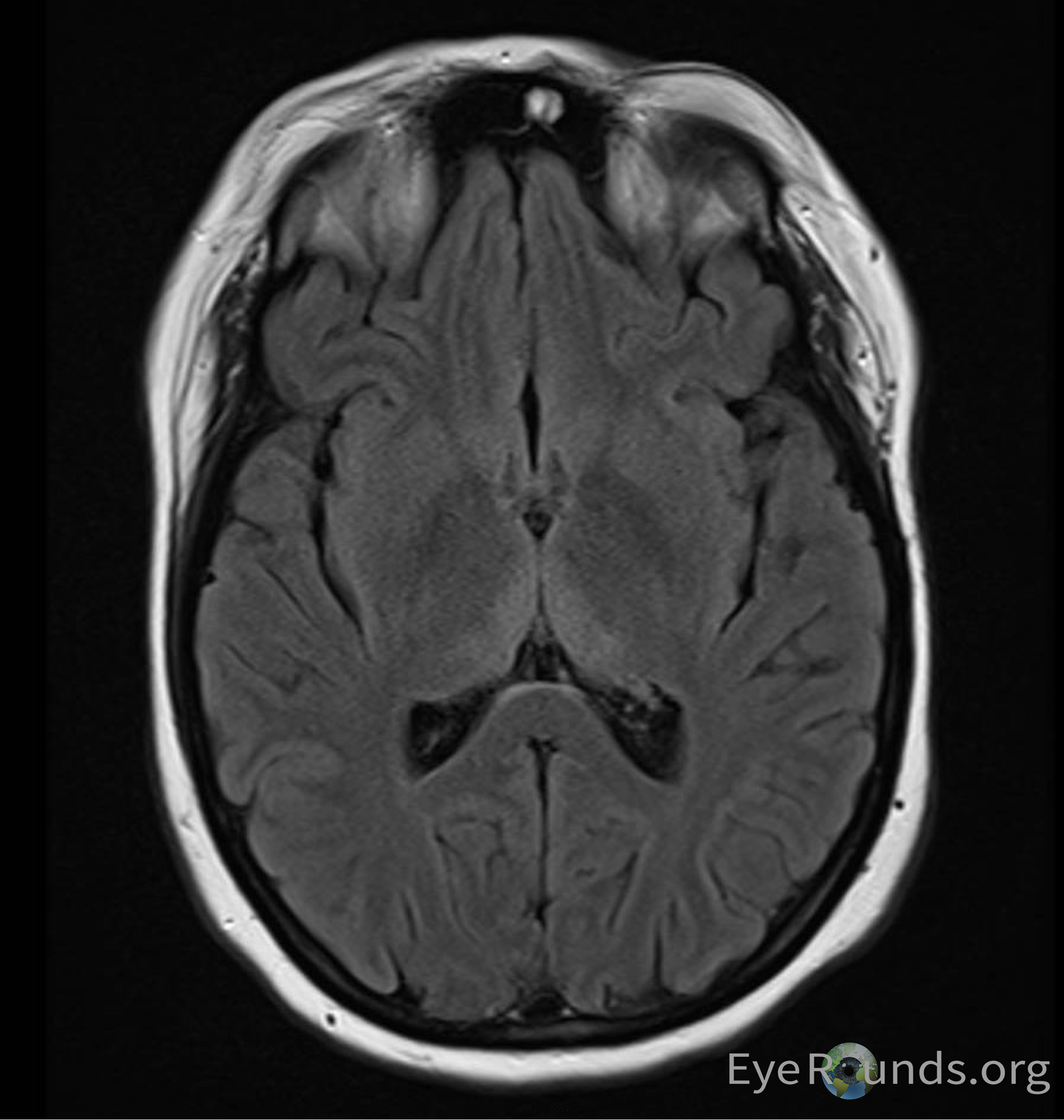
She underwent urgent lumbar puncture (LP) with an unremarkable cerebrospinal fluid analysis without evidence of inflammation or infection. Electromyogram and nerve conduction studies did not show evidence of demyelination. Anti-Gq1b antibodies were drawn and ultimately returned negative. MRI brain showed abnormal FLAIR signal in the medial thalami. Thiamine levels were drawn which eventually returned abnormally low.
Treatment was initiated with IV thiamine 500 mg three times daily for 9 total doses followed by PO repletion with 100 mg daily. She reported improvement of her lower extremity weakness after 4 days, paresthesia, numbness, and diplopia had improved by 4 months, with some residual oscillopsia even in primary gaze. After 1 year of PO repletion, she was symptomatic only in eccentric gazes.
After 4 days of IV thiamine the patient’s lower extremity weakness completely resolved. Neurological exam at the time of discharge was notable for decreased sensation of bilateral lower extremities, inability to abduct either eye, and nystagmus on primary, horizontal, and vertical gaze.
DIAGNOSIS: Wernicke Encephalopathy (WE)
DISCUSSION
Etiology/Pathophysiology
The prevalence of WE varies widely by geography. In the Western world brain lesions associated with WE are observed in between 0.4 and 2.8% of autopsies, and as much as 12.5% of those with Alcohol Use Disorder (AUD)(1 ,2). WE lesions have a prevalence as high as 59% in alcohol related deaths(3). Men are significantly more likely to misuse alcohol, and therefore, men are affected at higher rates(4). Interestingly, the female-to-male ratio for WE is higher than the ratio for alcohol dependence, suggesting that women are more susceptible to developing WE(5).
Thiamine deficiency and Wernicke’s Encephalopathy are a known complication of bariatric surgery. Abarbanel et al. examined 500 patients who underwent bariatric surgery. They reported that 23 (4.6%) of these patients experienced neurological complications, 9% of which (0.4% of total population) were specifically affected by WE(6). WE and other neurological complications of bariatric surgery appear to be directly related to severity of post-operative emesis and poor nutrition(7 ,8).
Pathophysiology
Thiamine is an important cofactor for several enzymes involved in glycolysis, the citric acid cycle, and the pyruvate phosphate shunt. It is absorbed in the duodenum in an energy-requiring active process; however, at extremely high (pharmacologic) concentrations, thiamine absorption is largely passive(9).
The body’s stores of thiamine are sufficient for 18 days after which the forementioned enzymes are impaired. This leads to pathognomonic brain lesions in regions that have particularly high thiamine turnover(10).
The first effects of thiamine deficiency manifest in astrocytes which require high concentrations of alpha-ketoglutarate dehydrogenase to metabolize the glutamate that functions as a neurotransmitter in the central nervous system (CNS). The accumulation of glutamate in astrocytes leads to cytotoxic edema. Later in the disease course pyruvate dehydrogenase is affected resulting in a large decrease in CNS glucose utilization.
In addition to dysfunction in glucose and glutamate metabolism, cells experience intense oxidative stress due to impairment of transketolase which functions to recycle NADPH and thus glutathione(11).
The intracellular accumulation of lactate and increased oxidative stress results in apoptosis and irreversible lesions affecting primarily the medial thalamus, mamillary bodies, and periaqueductal gray matter(12).
At autopsy, acute lesions demonstrate vascular congestion, microglial proliferation, and petechial hemorrhages. Eventually lesions will demonstrate demyelination and gliosis. Mamillary body atrophy is a specific indicator of chronic WE(13).
WE can present in anyone with a nutritional thiamine deficiency. Interestingly, those with alcohol use disorder (AUD) are at particularly high risk(14). The etiology is multifactorial. People with AUD typically have poor eating habits, sometimes going days without eating any nutrients, consuming only high carb beverages. In people with already low thiamine stores, these carb-dense meals further deplete thiamine levels through the metabolism of carbohydrates by thiamine dependent enzymes involved in glycolysis and the citric acid cycle, such as pyruvate dehydrogenase and alpha-ketoglutarate dehydrogenase(14). Secondly, alcohol damages the lining of the intestine, directly inhibiting absorption of thiamine as well as folic acid, which is thought to be involved in the transport of thiamine(15). Finally, chronic alcohol exposure down-regulates the activity of thiamine pyrophosphokinase, an enzyme responsible for converting thiamine to its active form(16).
Signs/Symptoms
The classic triad of WE are ataxia, encephalopathy, and oculomotor dysfunction. However, clinical suspicion should be high in any patient AUD who has any one of these symptoms(4).
Ataxia in WE is primarily due to involvement of the cerebellar vermis resulting in truncal ataxia and incoordination of gait.
The presentation of encephalopathy is variable in WE. Some patients will exhibit profound disorientation, whereas others will present with a milder encephalopathy detectable with cognitive testing demonstrating impaired memory and learning.
Finally, there are a variety of ocular manifestations in WE as described below.
Ocular Manifestations
The first, and most common ocular manifestation of WE is nystagmus. Typically consisting of a gaze-evoked horizontal component as well as a vertical, upbeat nystagmus (UBN) component; however, a downbeat nystagmus (DBN) can be elicited with special maneuvers, including positional changes, head shaking, and convergence. Most patients with these findings will develop a permanent DBN within one year of presentation.
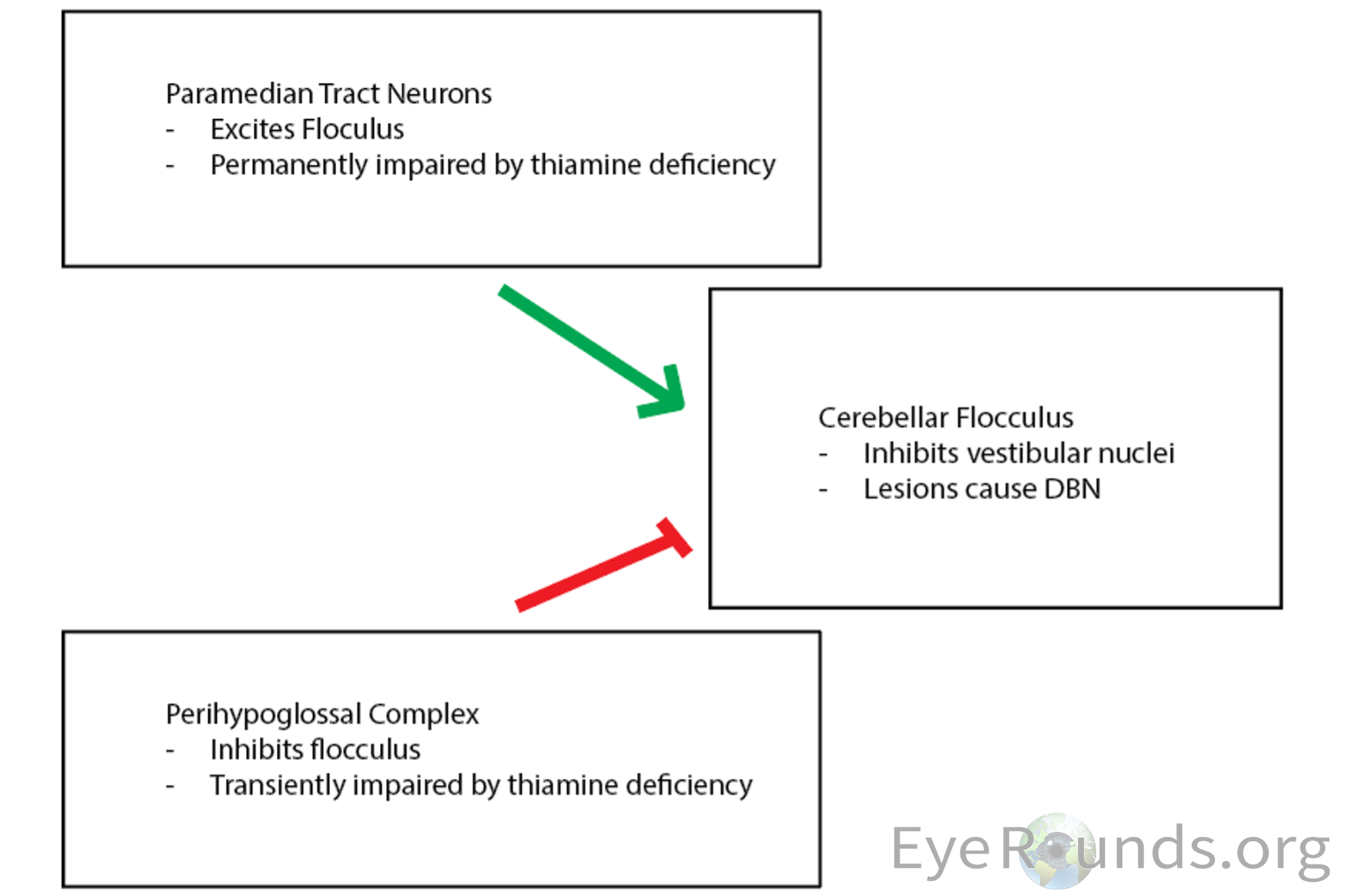
The exact pathophysiology of this UBN/DBN transition is not completely understood; however, Kattah et al. hypothesizes that these findings are closely related to the balance of inhibitory/excitatory signaling to the flocculus of the cerebellum(17). The flocculus projects to the brainstem and inhibits vestibular nuclei, which mediate upward slow phases. Therefore, lesions of the flocculus lead to an upward bias and DBN. Inhibitory signals are received by the flocculus from a collection of nuclei in the perihypoglossal complex of the caudal medulla. These nuclei are believed to be transiently impaired by thiamine deficiency. This disinhibition of the flocculus results in an UBN. The transition to DBN is explained by permanent impairment in the paramedian tract neurons (A collective term referring to clusters of neurons scattered along the midline of the pons and medulla, which project to the cerebellum) which typically function to excite the flocculus. Long-term thiamine deficiency is believed to cause permanent dysfunction of paramedian tract neurons, ultimately resulting in DBN.
The next most common ocular manifestation of WE are bilateral abducens palsies followed closely by conjugate gaze palsies. Internuclear ophthalmoplegia has been reported but is a relatively rare finding. These symptoms are a consequence of WE lesions affecting the pontine tegmentum as well as the abducens and oculomotor nuclei(18 ,19).
WE can also manifest with vestibulo-ocular findings. This presents as failure to maintain gaze with the head impulse test (HIT)(20). HIT exam will reveal disruption of the horizontal vestibulo-ocular reflex. Interestingly, the vertical vestibulo-ocular reflex is spared suggesting selective impairment of the medial vestibular nuclei(21 ,22).
Finally, though rare, fundus changes have also been described in WE. Findings include optic disc edema and retinal hemorrhages(22). These signs are described in only 2-4% of WE patients(22). If severe enough optic neuropathy with disc edema may progress to vision loss.
Testing/Laboratory work-up
WE is primarily a clinical diagnosis that should be considered in any patient with poor nutrition or altered absorption of nutrients. The diagnosis can be confirmed with laboratory testing of blood thiamine (prior to supplementation) and red blood cell transketolase activity; however, treatment should not be delayed for test results.
Imaging
MRI lacks sensitivity but is a valuable method to confirm a diagnosis of WE. MRI demonstrating a symmetrically increased T2 signal in paraventricular regions of the thalamus, hypothalamus, mamillary bodies, periaqueductal region, floor of the fourth ventricle and middle cerebellum is 93% specific for WE(23).
Treatment/Management/Guidelines
WE should be treated promptly with thiamine supplementation. Clinical workup should not delay treatment. There are no randomized clinical trials to determine the exact dosing of IV thiamine in WE patient’s; however, IV thiamine is considered safe and can be reasonably administered according to a regimen of 500 mg infused over 30 minutes, 3 times daily for 2 days and then 250 mg once daily for 5 days(24).
It is critical to administer thiamine prior to any glucose containing fluids or nutrition, as the metabolism of glucose is dependent on thiamine. Administration of glucose in a patient with WE prior to thiamine can result in worsening neurologic dysfunction, as astrocytes of the central nervous system are particularly dependent on thiamine to metabolize the glutamate used as a neurotransmitter in the CNS. Furthermore, pyruvate dehydrogenase is dependent on thiamine as a co-factor. Without the function of this critical enzyme the products of glycolysis are shunted towards the production of lactate. The accumulation of lactate and glutamate within astrocytes can acutely worsen cytotoxic edema(10).
These patients are rarely deficient in exclusively thiamine, often necessitating repletion of other B-vitamins and electrolytes(25).
Patients who experience WE should adhere to daily oral thiamine supplementation until they are no longer considered at risk of deficiency.
EPIDEMIOLOGY OR ETIOLOGY
|
SIGNS
|
SYMPTOMS
|
TREATMENT/MANAGEMENT
|
Chenoweth D, Ahmed SB, Linton EF. Wernicke Encephalopathy. EyeRounds.org. July 16, 2024. Available from https://EyeRounds.org/cases/358-wernicke-encephalopathy.htm

Ophthalmic Atlas Images by EyeRounds.org, The University of Iowa are licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License.

Address
University of IowaLegal
Related Links