INITIAL PRESENTATION
Chief Complaint: Congenital nystagmus and early-onset progressive myopia
History of Present Illness:
A 4-year-old male with congenital nystagmus presented for evaluation of reported decreased vision since early infancy and persistent nystagmus. The patient received his first pair of eyeglasses at approximately 2 years old for high myopia and wore his eyeglasses consistently. His vision appeared somewhat improved while wearing the eyeglasses. His parents noted that his nystagmus seemed less obvious when wearing his eyeglasses. Additionally, his parents reported that he displayed light sensitivity in bright lighting conditions. He did not have strabismus.
Past Ocular History:
Past Medical History:
Medications:
Allergies:
Family History:
Social History:
OCULAR EXAMINATION
OD and OS:
OD and OS:
| Wearing Rx | |||
|---|---|---|---|
| Sphere | Cylinder | Axis | |
| Right | -8.00 | +3.50 | 096 |
| Left | -8.00 | +3.00 | 079 |
| New Prescription | |||
| Sphere | Cylinder | Axis | |
| Right | -9.00 | +3.50 | 096 |
| Left | -9.00 | +3.00 | 079 |
DIFFERENTIAL DIAGNOSIS:
CLINICAL COURSE
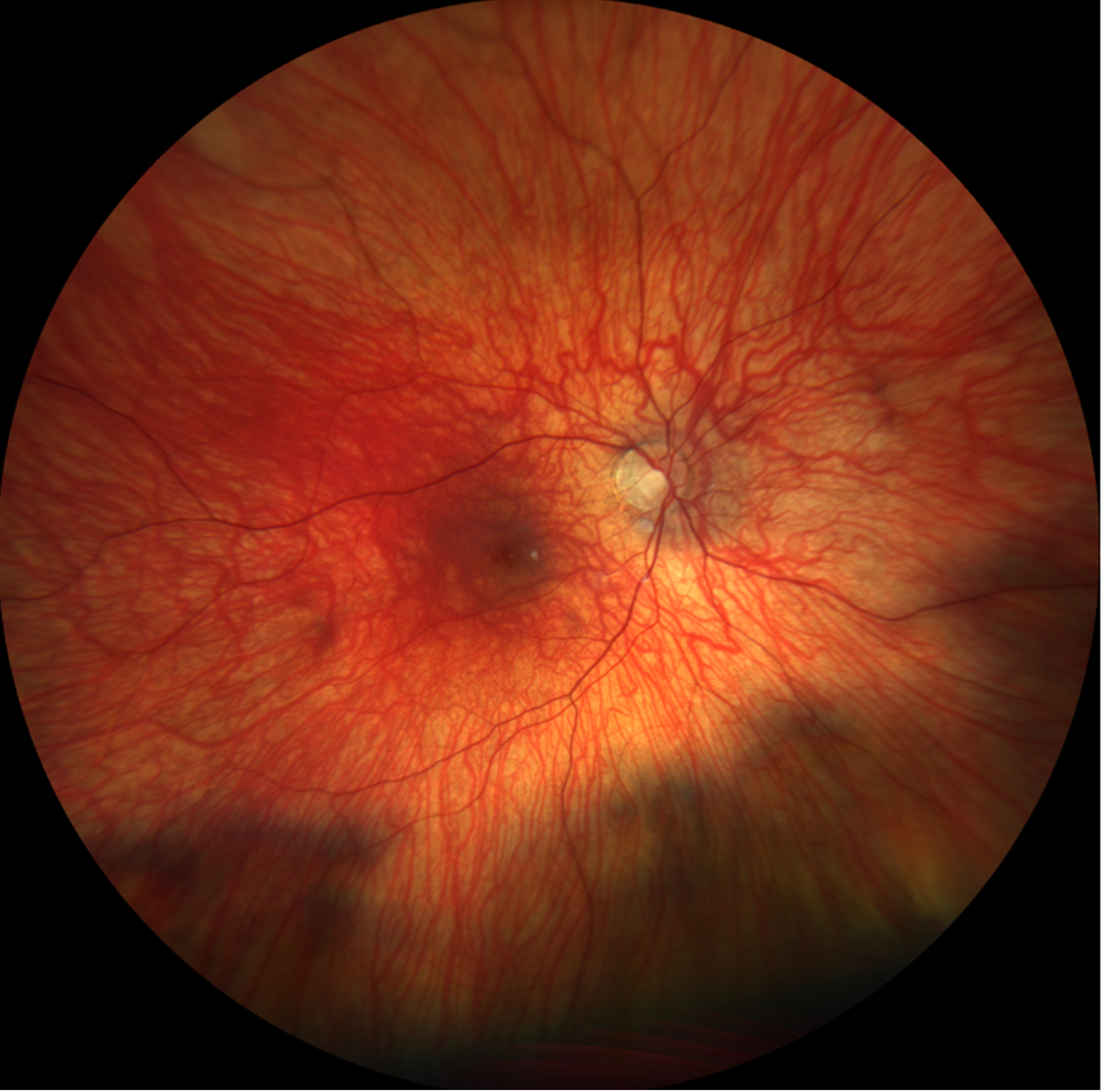
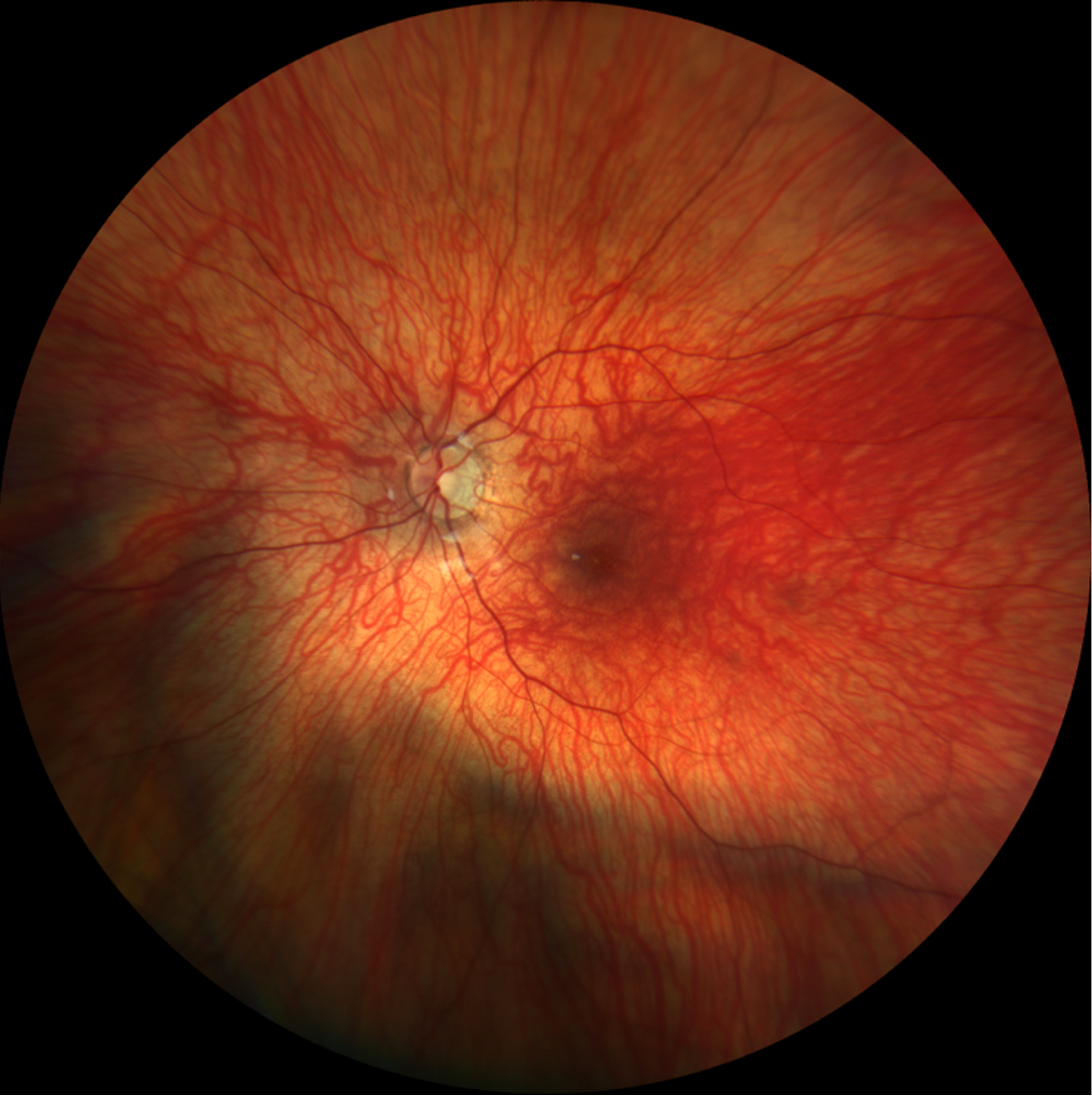
Given his clinical presentation and relevant testing results, blue cone monochromacy was highest on the differential diagnosis. Specific findings consistent with this diagnosis included his congenital nystagmus, decreased best-corrected visual acuity, moderate light sensitivity, family history of a maternal grandfather with poor vision (suggestive of possible x-linked recessive inheritance pattern), decreased amplitudes in light-adapted full field ERG, early-onset high myopia, and decreased color vision on Ishihara test with relatively normal fundus appearance.
Prior to being seen in our clinic, a previous provider was concerned for retinal dystrophy and ordered a sponsored inherited retinal dystrophy panel, which revealed multiple genetic variants but no definitive explanation of the phenotype. The findings were:
The genetic testing showed that the patient was negative for pathogenic variants in genes associated with Achromatopsia and did not have any variants of unknown significance in those genes.
Based on these results, and the clinical and electrophysiological findings, we determined that additional genetic testing was necessary to clarify the underlying etiology of his vision. Targeted genetic testing showed that the patient’s father possessed the GUCY2D VUS with no associated phenotype except red/green dyschromatopsia. Genetic testing for the cone opsin genes (OPN1LW and OPN1MW) responsible for Blue Cone Monochromacy revealed that the patient has a hemizygous pathogenic variant in the OPN1MW gene c.607T>C (p. Cys203Arg). Maternal testing confirmed that the mother was a carrier of the disease. This result explained his personal and family history of vision concerns as well as his nystagmus, dyschromatopsia, and decreased vision. We reviewed his genetic testing information and implications for the family in addition to discussing implications of the X-linked inheritance pattern. Pre- and post-test genetic counseling was provided to the family.
DIAGNOSIS: Blue Cone Monochromacy
DISCUSSION
Blue cone monochromacy (BCM) is a rare, X-linked congenital cone dystrophy characterized by impaired color discrimination, early-onset high myopia (in many but not all affected individuals), congenital nystagmus, and photophobia. (1) Patients typically have reduced visual acuity from early infancy, with symptoms slowly progressing into adulthood. Many of the presenting features overlap with other cone dysfunction disorders such as achromatopsia or rod monochromatism. (1)
BCM affects an estimated 1 in 100,000 individuals. (1) Since it is inherited in an X-linked recessive pattern, it primarily affects males, while female carriers are typically asymptomatic. The disease occurs worldwide without significant racial or ethnic predilection. The condition results from pathogenic variants affecting the OPN1LW and OPN1MW opsin gene clusters on the X chromosome, which encode the red (L-cones) and green (M-cone) photopigments, leading to a reliance on rod photoreceptors and blue cones (S-cones) for visual processing. (1,2) Since rod photoreceptors are more sensitive in low-light conditions, patients may experience difficulties in bright environments due to photophobia. The inability to detect red and green wavelengths manifests as severe color vision deficits on standard testing, such as the Ishihara test. (1)
For this patient and many others who have BCM, high myopia was the presenting feature prompting referral. High pediatric myopia warrants a thorough workup to evaluate for an underlying cause or association, such as this case. With emerging advancements in myopic control strategies, it is important to identify and treat high myopic patients as early as possible. The efficacy of current methods for slowing myopia progression is unknown for myopia-associated retinal dystrophy.
In BCM, high myopia can exacerbate visual impairment and increase the risk of complications. (3) Regular monitoring for myopic retinal degeneration is essential to prevent complications such as retinal detachment which can lead to permanent vision loss. (4) Additionally, patients and their guardians should be advised on the risks of contact sports in pediatric patients and recommend the use of sport goggles for protection in extracurricular activities. (4)
Genetic testing plays a critical role in the diagnosis and confirmation of inherited retinal diseases such as BCM. However, the results of any genetic test must be interpreted in the context of a well-defined phenotype. Ordering broad or non-targeted genetic panels without prior phenotypic characterization such as a detailed clinical exam, electrophysiology studies, and OCT can lead to ambiguous or uninterpretable results. A precise clinical workup not only informs genetic panel and/or test selection but also ensures that identified variants are meaningful. Without proper phenotyping, VUS may lead to diagnostic confusion and unnecessary investigation into conditions that are inconsistent with the patient’s presentation, potentially delaying accurate diagnosis and management while creating anxiety for patients and families and wasting resources.
It is equally important to understand the scope and limitations of the genetic test being used, including which genes are covered in a panel and which are omitted. Understanding whether the test detects structural variants, copy number changes, or complex rearrangements in gene clusters will help guide one’s use and interpretation of genetic testing. In our patient, a broad Inherited Retinal Dystrophy panel was initially ordered. Although this panel revealed multiple VUS, none of the findings adequately explained the patient’s full phenotype. Notably, genes associated with BCM were not included in the original panel. Given the high clinical suspicion for BCM based on phenotypic and electrophysiologic findings, we pursued targeted testing, which ultimately confirmed a diagnosis of BCM. A phenotype-first approach not only prevents detection and possible misinterpretation of VUS, but also helps clinicians efficiently arrive at an accurate diagnosis through targeted testing.
Testing/Diagnostic Work-up:
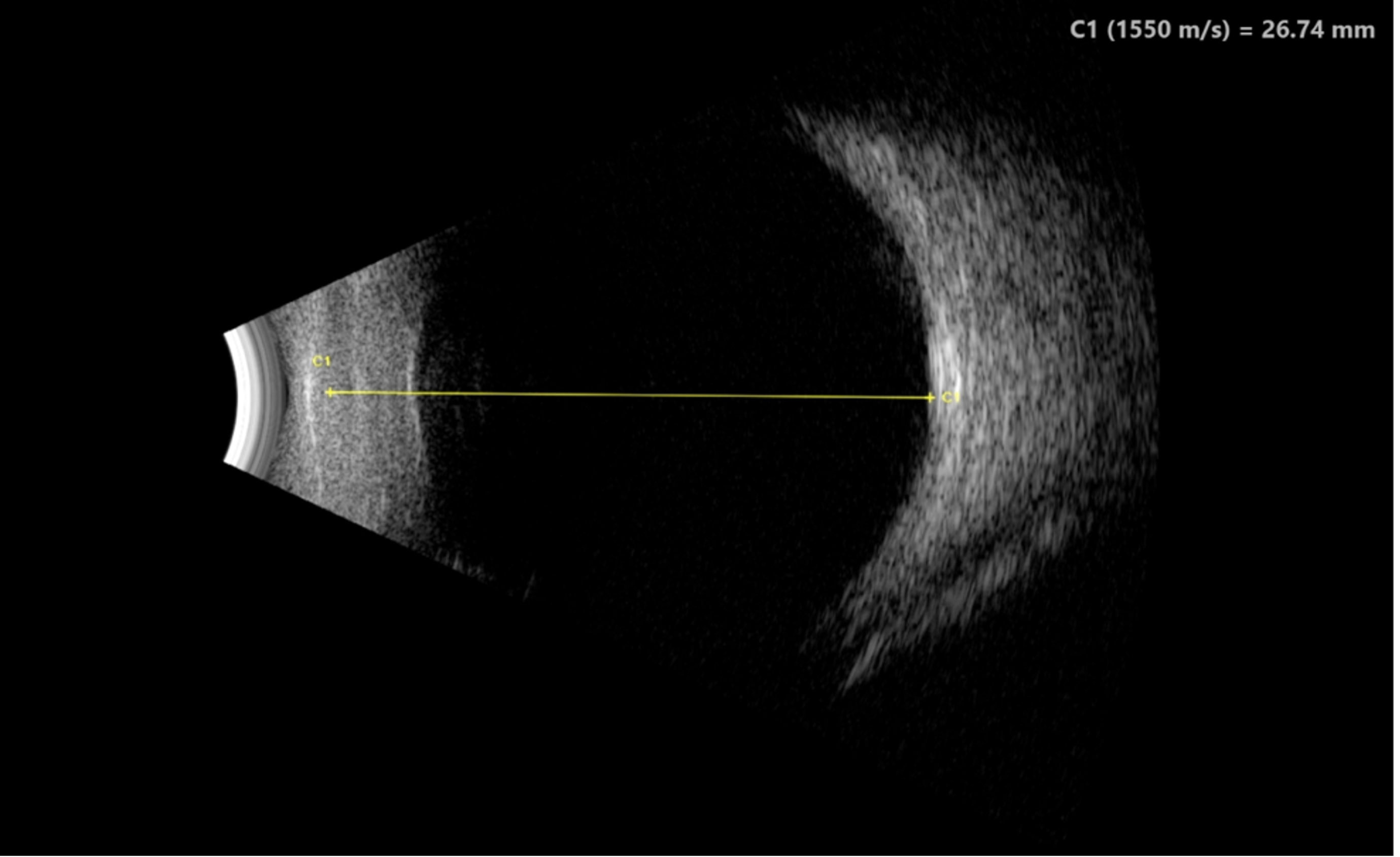
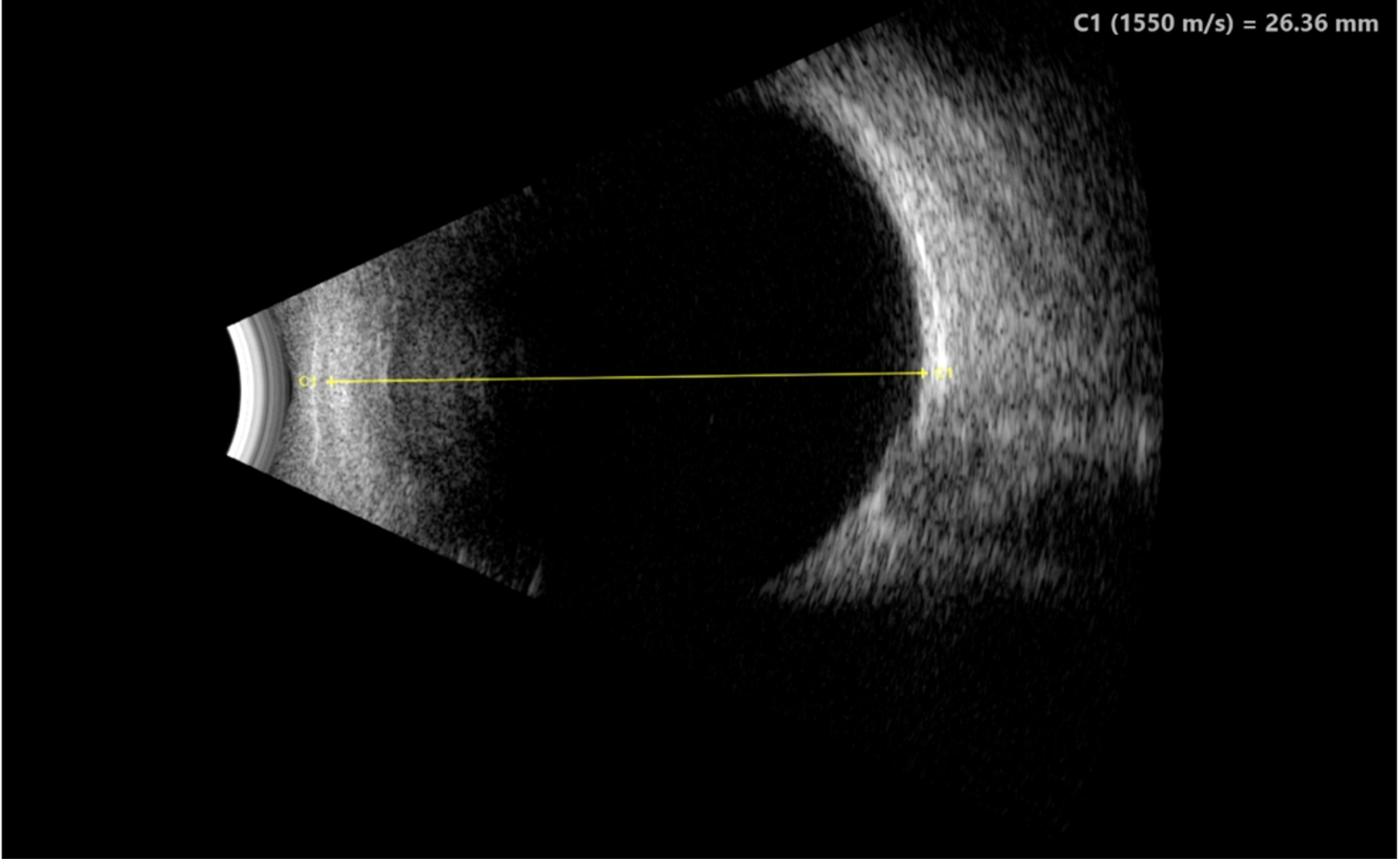
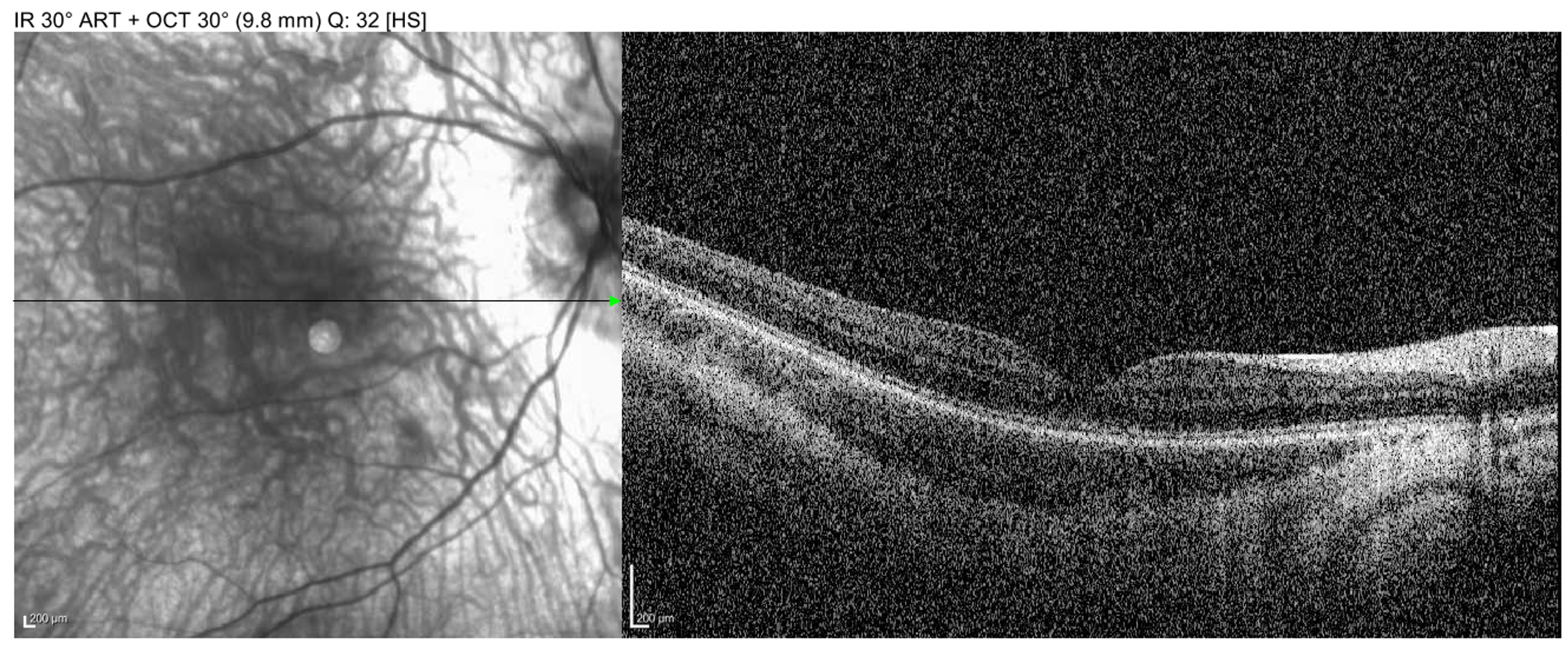
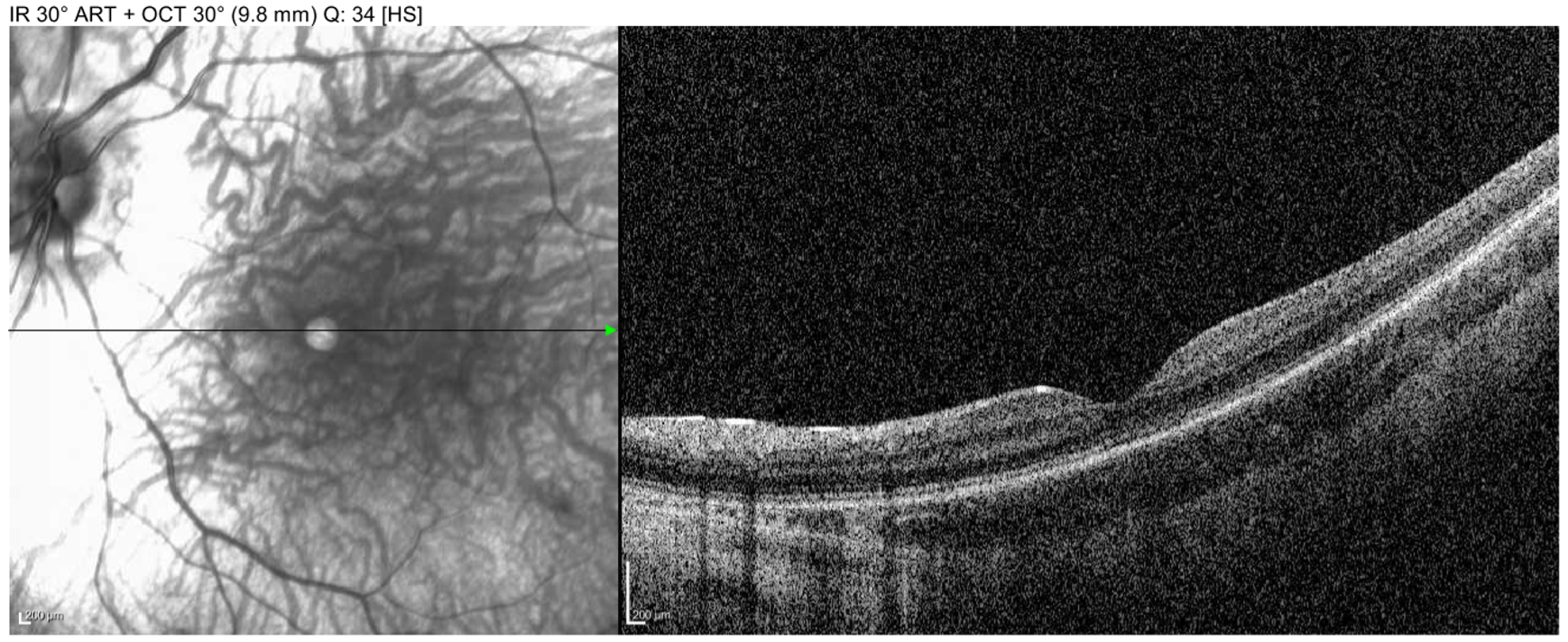
Imaging:
Treatment/Management/Guidelines:
Overall, BCM typically has slow or very little progression over time. (7,8) In infancy, patients present with congenital nystagmus and visual acuity is somewhat reduced. As children become older, photophobia and reduced color vision become more evident, nystagmus may regress, and decreased visual acuity becomes more apparent. (7,8) Vision typically ranges from 20/80 to 20/200. As patients transition to adulthood, their visual acuity is expected to remain decreased but in a functional range. Nevertheless, most patients are unable to drive or complete tasks that require fine visual acuity. (7,8)
There currently is no cure for BCM, but management focuses on maximizing visual function and quality of life through corrective lenses, tinted lenses, visual aids such as magnifiers, and educational support such as an individualized education program (IEP) for children in school. Long-term prognosis varies, but most individuals maintain stable visual acuity into adulthood. Genetic counseling is crucial to discuss inheritance patterns and potential implications for family members.
As genetic testing for the disease has become more widespread, so has research into treatment options for patients with BCM. Currently, two different mouse model therapies are being studied to improve vision in BCM patients, gene augmentation, and gene editing. Gene augmentation consists of delivering a functional copy of the gene to the host. In contrast, gene editing directly edits and corrects the pathogenic variant in the patient. (3) Currently these models have proven to have utility solely in mouse models, yet they provide hope for potential future gene therapy in humans.
EPIDEMIOLOGY/ETIOLOGY
|
SIGNS
|
SYMPTOMS
|
TREATMENT/MANAGEMENT
|
Davis K, Noble PM, Dumitrescu AV. Blue Cone Monochromacy. EyeRounds.org. August 11, 2025. Available from https://EyeRounds.org/cases/369-blue-cone-monochromacy.htm

Ophthalmic Atlas Images by EyeRounds.org, The University of Iowa are licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License.

Address
University of IowaLegal
Related Links