Chief Complaint: 4 month old boy with a "blur" in one eye since birth according to his mother.
History of Present Illness: At 2 months of age, his eyes would "wander off." At 4 months, a local MD noted poor red reflex in left eye. The child was subsequently referred to pediatric ophthalmologist.
PMH/FH/POH: Normal spontaneous vaginal delivery at 38 weeks. FH non-contributory.
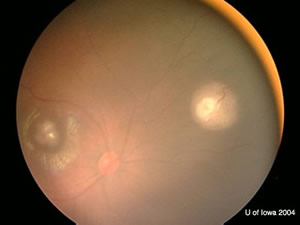 | 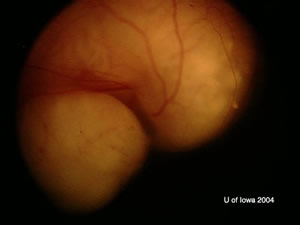 |
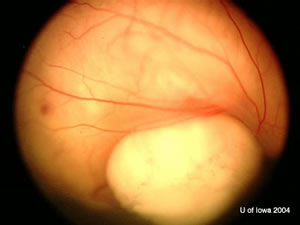
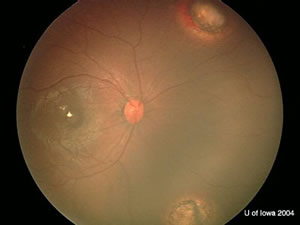
| OD: Multiple tumors in the posterior pole. | OS: Amelanotic large endophytic mass. |
 |
OD: 3 lesions, one in the macula with choroidal shadowing.
OS: Large, multilobulated calcified lesion with maximum height of 9 mm and choroidal shadowing.
No extraocular extension of mass
 |
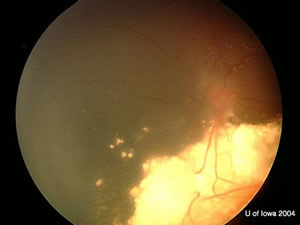 |
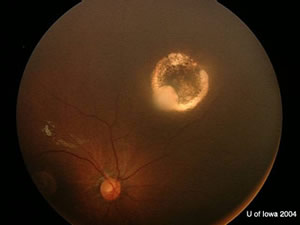
| OD. One month after chemoreductive therapy. | OS. One month after chemoreductive therapy. Left eye was removed later. |
 |
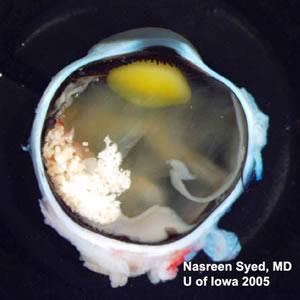
Gross Photo of Left Eye
|
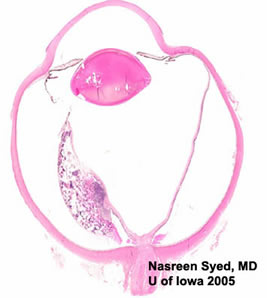
Anterior-Posterior section through the eye
|
H&E Stain
|
H&E Stain
|
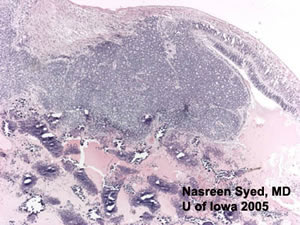
Flexner-Wintersteiner & Homer-Wright Rosettes
|
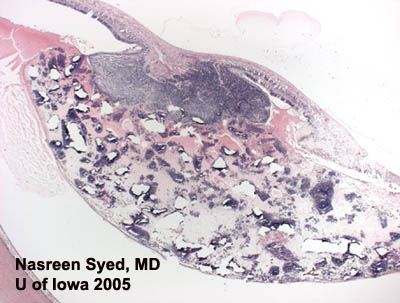
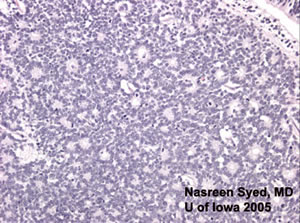
Islands of blue cells in a sea of necrosis are typical of retinoblastoma. The bottom left panel shows both types of rosettes: Flexner-Wintersteiner rosettes (with central lumens) and Homer-Wright rosette (eosinophillic centers). |
Retinoblastoma is the most common intraocular malignancy in children. Fortunately, 95% of children with retinoblastoma survive (because of early detection). The gene is located on the long arm of chromosome 13. The intact gene protects against expression of retinoblastoma. Knudson proposed that "2 hits" were necessary to eliminate both alleles and therefore inhibit tumor suppression.
Ocular signs include leukocoria, strabismus, uveitis, hyphema, iris neovascularization, or a transparent lesion in the neurosensory retina.
Other ocular disorders in children can present similar to retinoblastoma. The most common is persistent hyperplastic primary vitreous followed by Coats’ disease, ocular toxocariasis and ROP. 1 Diagnosis is usually obtained by an experienced observer due to its classic appearance. Ultrasonography is able to show calcium within the mass.
The number one goal in approaching treatment in patients with retinoblastoma is patient survival, followed by globe salvage then visual acuity. Treatment options include chemoreduction therapy, focal therapies alone, enucleation and external beam radiation. Chemoreduction therapy has emerged as an important initial approach to retinoblastoma treatment. The use of carboplatin, etoposide, and vincristine has been shown to shrink the size of the tumor allowing focal treatments such as cryotherapy, focal laser therapy, or thermotherapy to further preserve vision and avoid enucleation. Recent studies have shown a decrease of 35% of tumor base and 50% decrease of tumor thickness after 2 cycles. Ocular salvage rates have improved as well.
The two major concerns associated with treating with external beam radiation are the germline alteration of the RB gene producing an increase risk of secondary malignancies (osteosarcoma) and the risk of side effects of radiation on the eye (retinopathy, neuropathy and cataract).
Histologic specimens of retinoblastoma show cells with round, oval nuclei that are twice the size of lymphocytes. As tumor extends into the vitreous and outgrows its blood supply, a characteristic pattern of "islands of blue cells in a sea of necrosis" is formed. (Figure 4)
A feature of retinoblastoma includes the formation of Flexner-Wintersteiner rosettes which are formations of retinal differentiation. (See Figure 4) The cells of these rosettes surround a central lumen. Another pattern seen in retinoblastomas is the Homer-Wright rosette where the rosette is filled with an eosinophilic substance. These rosettes are also seen in other neuroblastic tumors.
EPIDEMIOLOGY
Genetics
|
SIGNSMAJOR COMPONENTS
|
SYMPTOMSLife threatening problems1. Metastasis: within first year Risk factors for metastasis:
2. Trilateral retinoblastoma
3. Secondary Malignancies
|
TREATMENTTreatment Goals:
Options:
|
Coombs J, Boldt HC. Retinoblastoma: 4 month old boy with a "blur" in one eye since birth according to his mother. EyeRounds.org. February 21, 2005; Available from: http://www.EyeRounds.org/cases/case27.htm.

Ophthalmic Atlas Images by EyeRounds.org, The University of Iowa are licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License.

Address
University of IowaLegal
Related Links