
An 18-year-old male presented to clinic for regular monitoring of ocular manifestations of methionine synthase deficiency type homocystinuria, specifically cobalamin deficiency of the Cb1G subtype. He complained of some photophobia and intermittent inward turning of his eyes. Additionally, he was noted to have nystagmus and subnormal vision.
The patient was first evaluated by ophthalmology at four months of age at which time he was noted to have large amplitude horizontal pendular nystagmus and poor fixation. His dilated fundus exam (DFE) showed indistinct foveal reflexes and retinal pigment epithelium (RPE) granularity. Several months later, the patient was re-examined and noted to have high astigmatism in both eyes (OU) for which he was given glasses. At about one year of age an electroretinogram (ERG) was performed, which showed generalized reduction. Over time, he developed an esotropia of 40-50 prism diopters and mild amblyopia in the left eye (OS). He ultimately underwent bilateral medial rectus recessions of 5.5 mm at six years of age.
At age 11 years, his best corrected vision was noted to be 20/50 in the right eye (OD) and 20/60 OS. He had progressive myopia and astigmatism, especially OS. However, no ectopia lentis was detected. Fundus examination demonstrated yellowish flecks in the maculae OU, which were suspicious for Stargardt disease. A workup for hereditary retinal degeneration was performed and was remarkable for just one mutation in ABCA, which was not diagnostic for autosomal recessive Stargardt disease.An ERG was repeated at age 16 due to subnormal vision which showed abnormal cone function. Optical coherence tomography (OCT) at the time showed loss of foveal depression and parafoveal outer retinal atrophy.
Additionally, the patient has multiple systemic manifestations of homocystinuria, including developmental delay, megaloblastic anemia, seizures, and chronic kidney disease. Despite early intervention in a metabolic program, including dietary restriction and prescription of betaine, his homocysteine levels were never well-controlled.
Distance (Snellen):
Manifest Refraction:
Color Vision (Ishihara):
(Tonopen)
 |
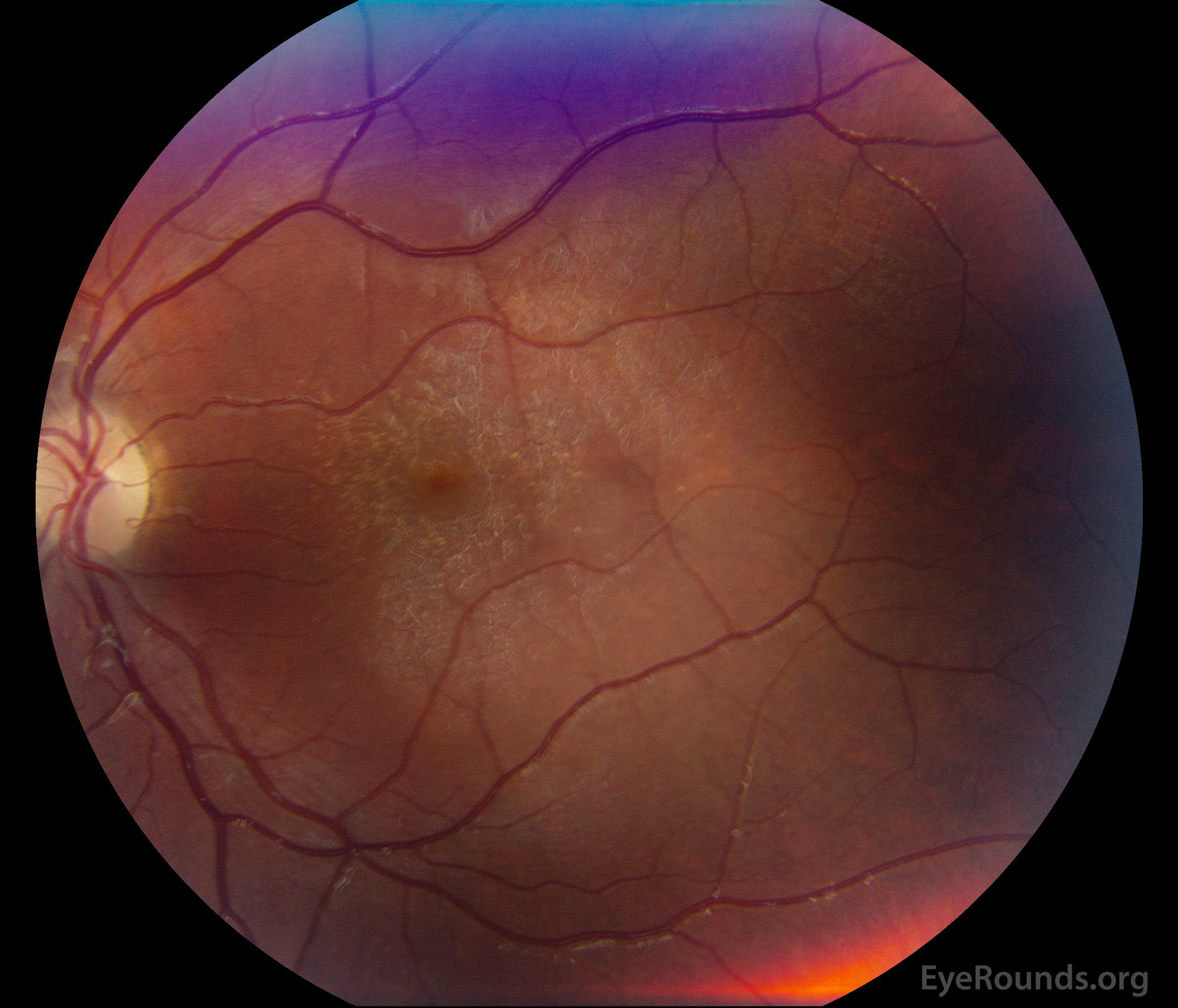 |
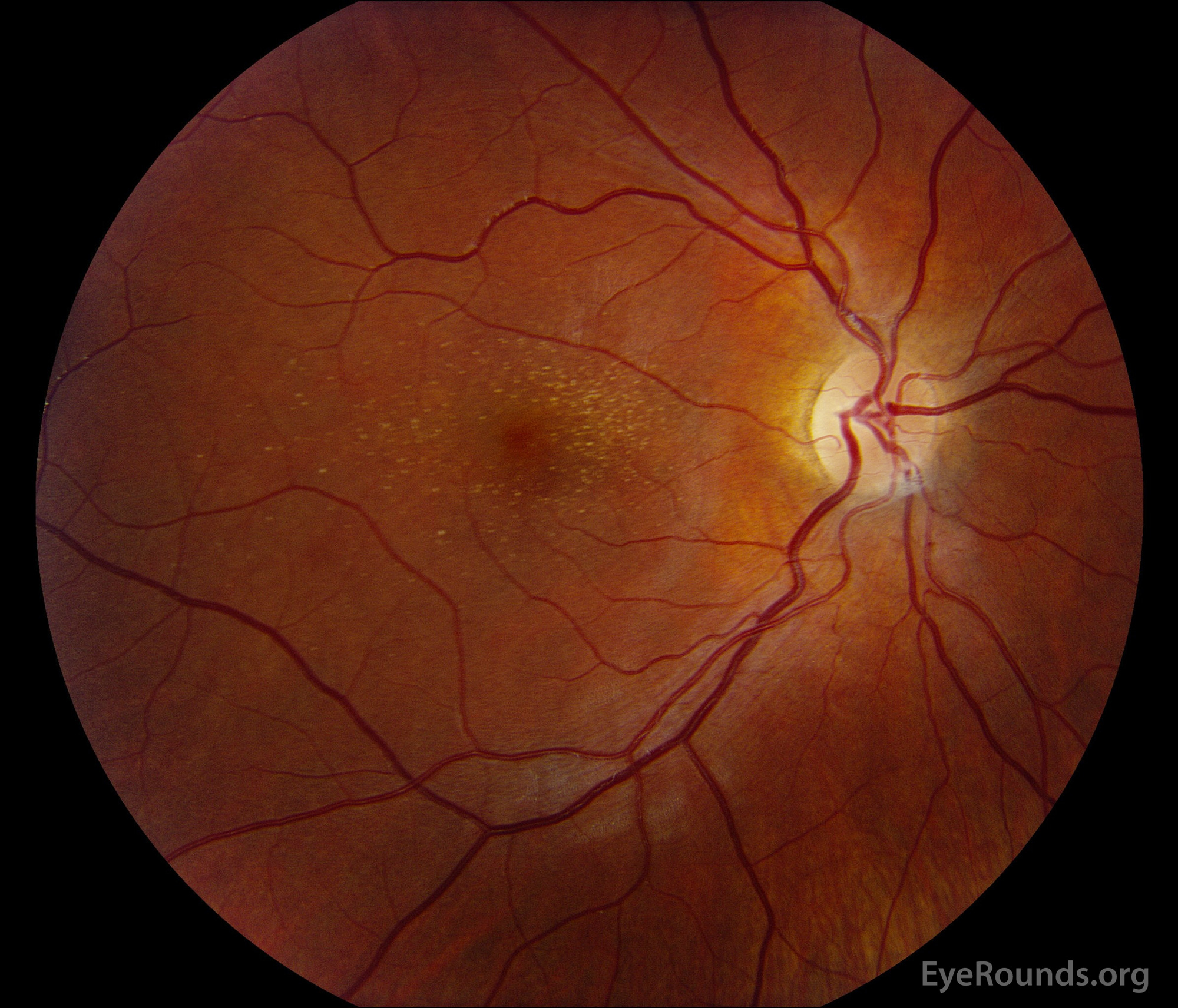
Figure 1: A, B. Color fundus photographs OD (A) and OS (B) show whitish flecks in the maculae.
Additional testing
 |
 |
 |
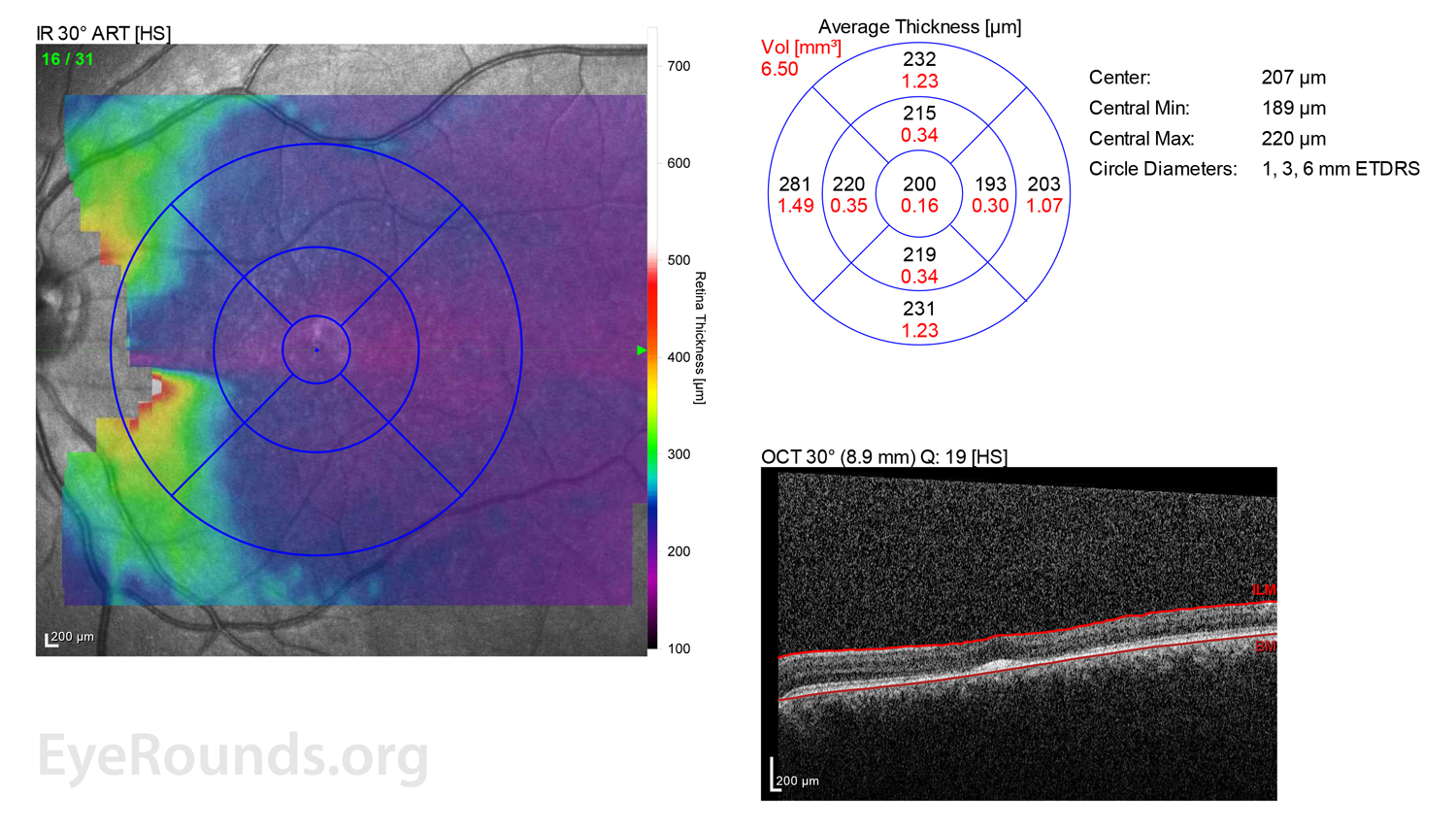 |
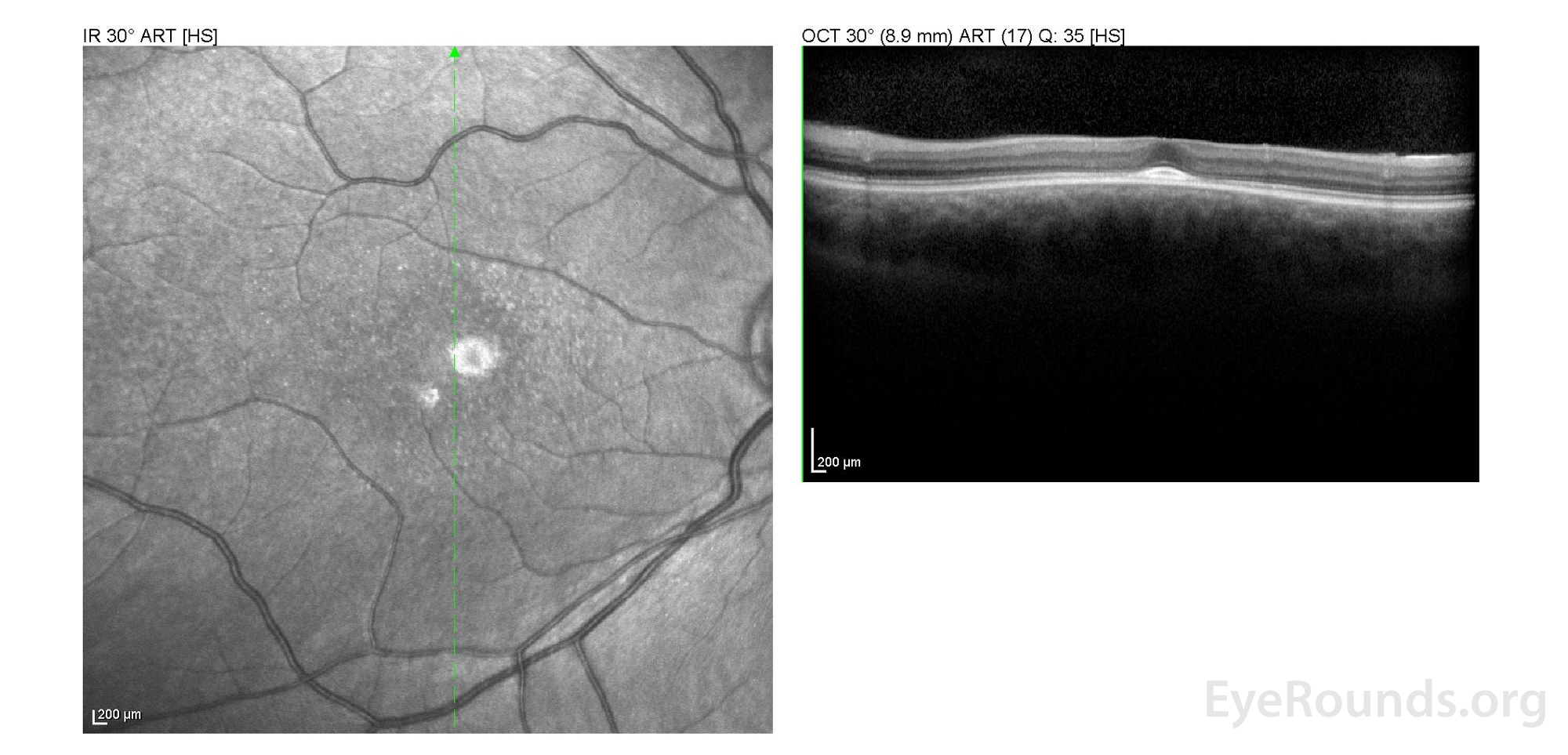
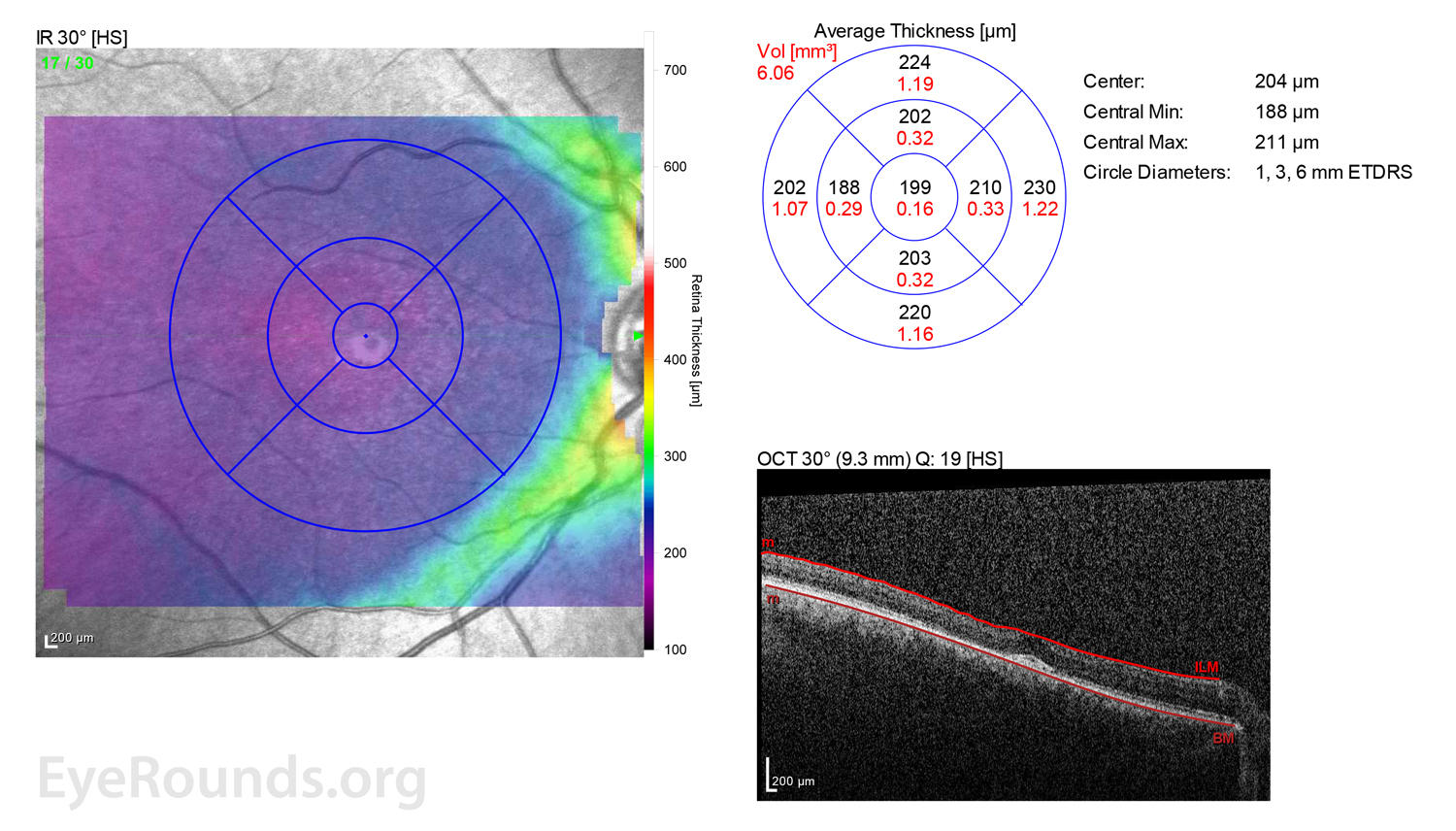
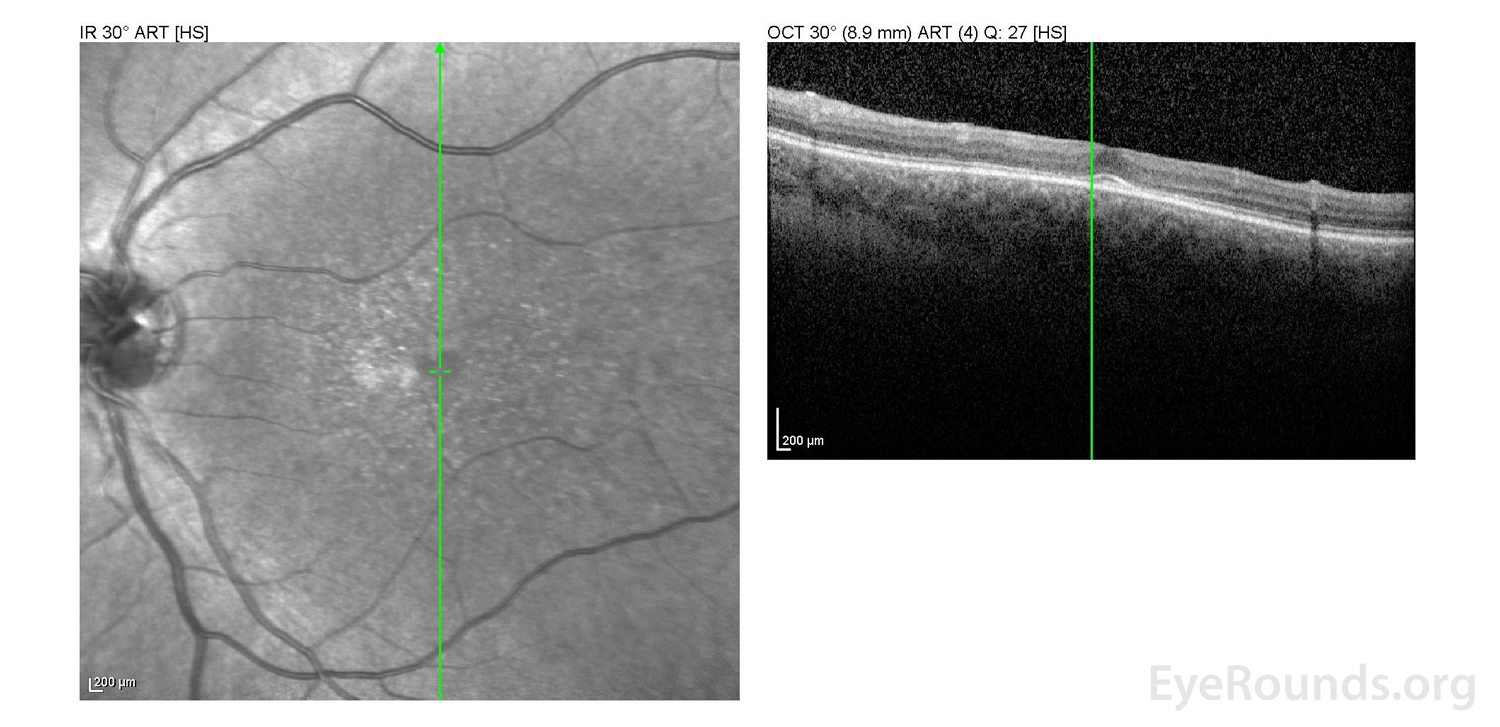
Figure 2: A, B. OCT OD (A) and OS(B) show diffuse retinal thinning with parafoveal atrophy of the outer retinal layers. There is an area of subfoveal hyperreflective foci OU.
 |
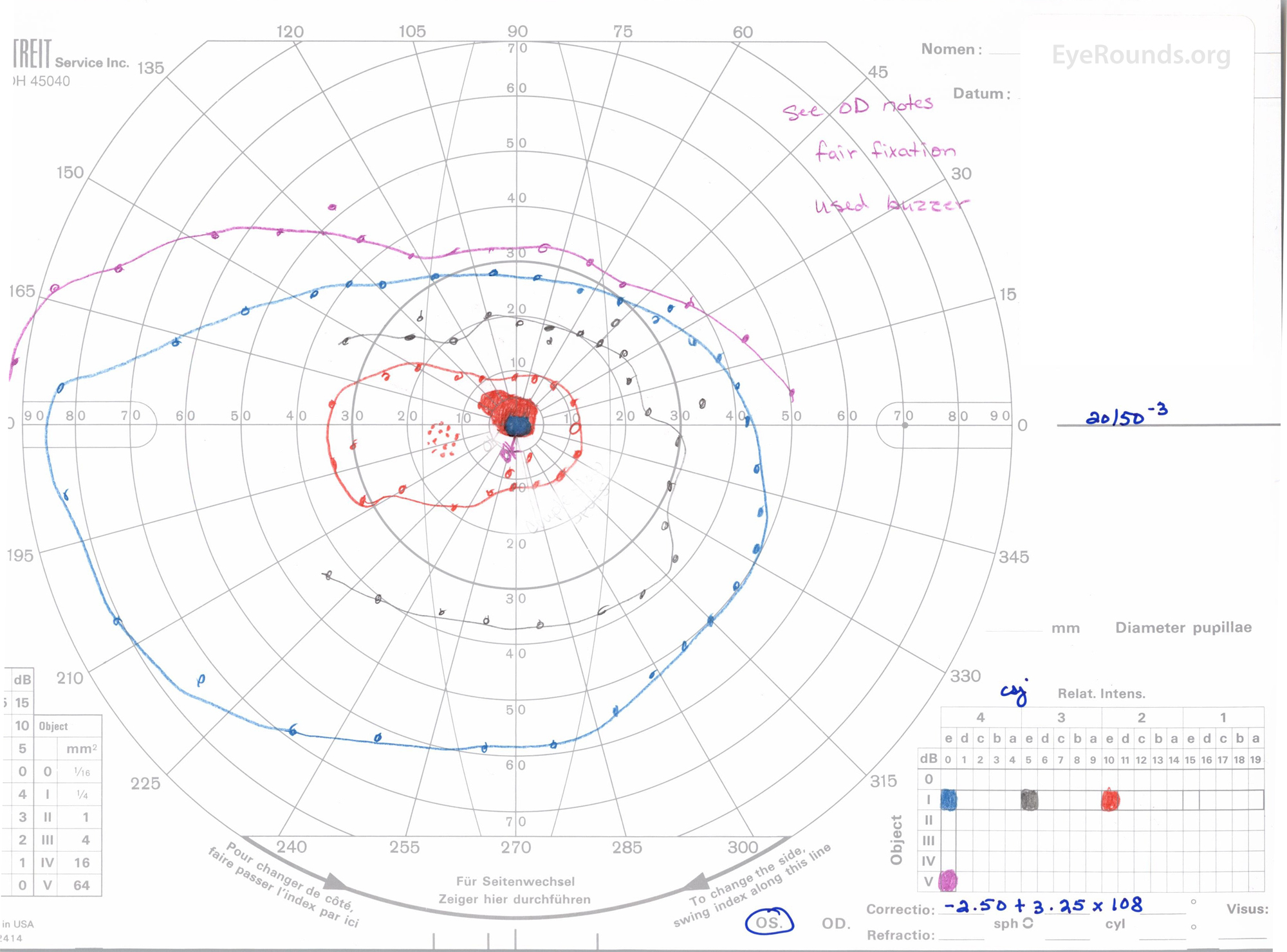 |
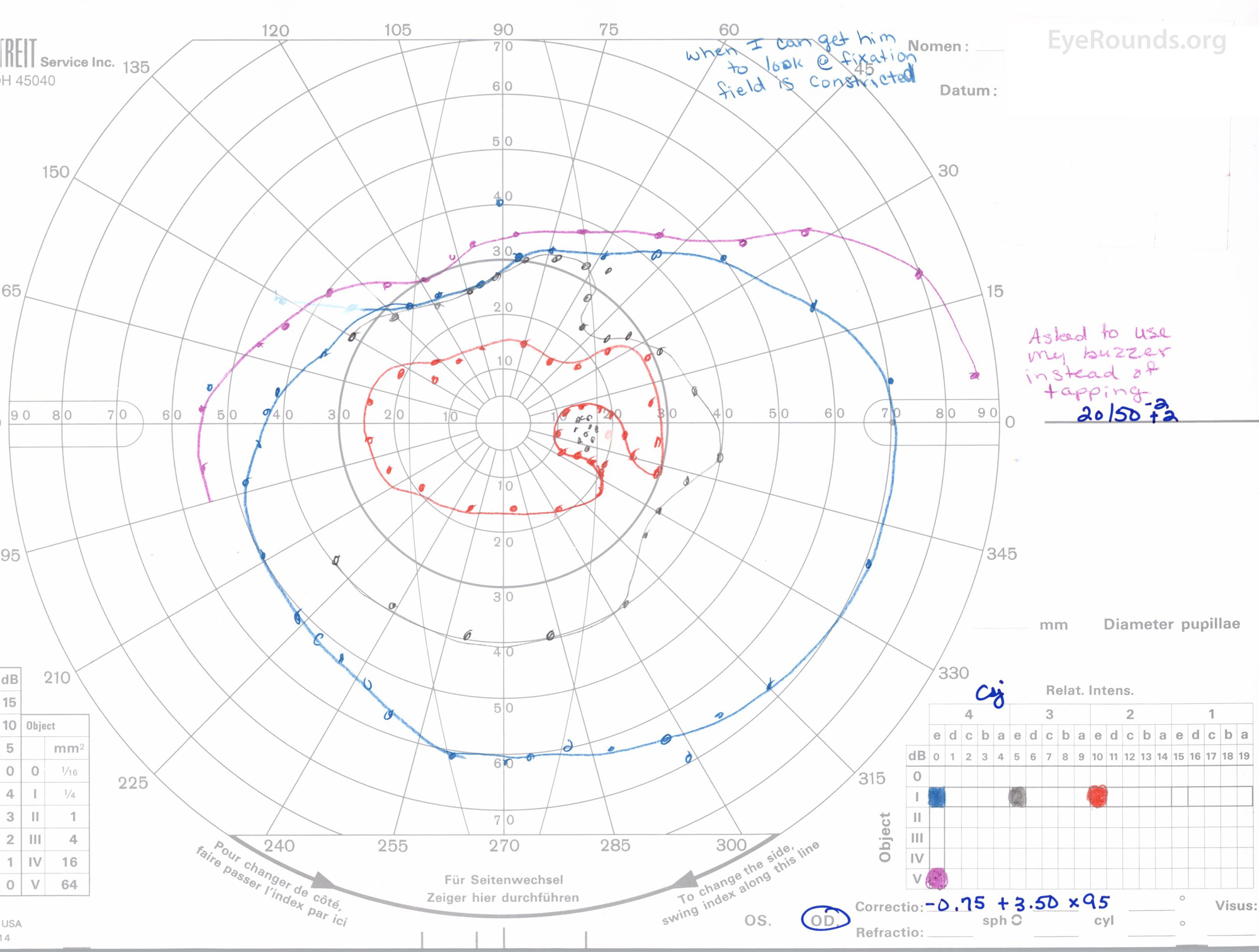
Figure 3: A, B. Goldmann Visual Field (GVF) OD (A) shows an overall full visual field to all isopters tested. GVF OS (B) shows a 5 degree central scotoma to the I4e isopter centrally and the I2e isopter peripherally but otherwise a full visual field to all isopters tested.
 |
 |
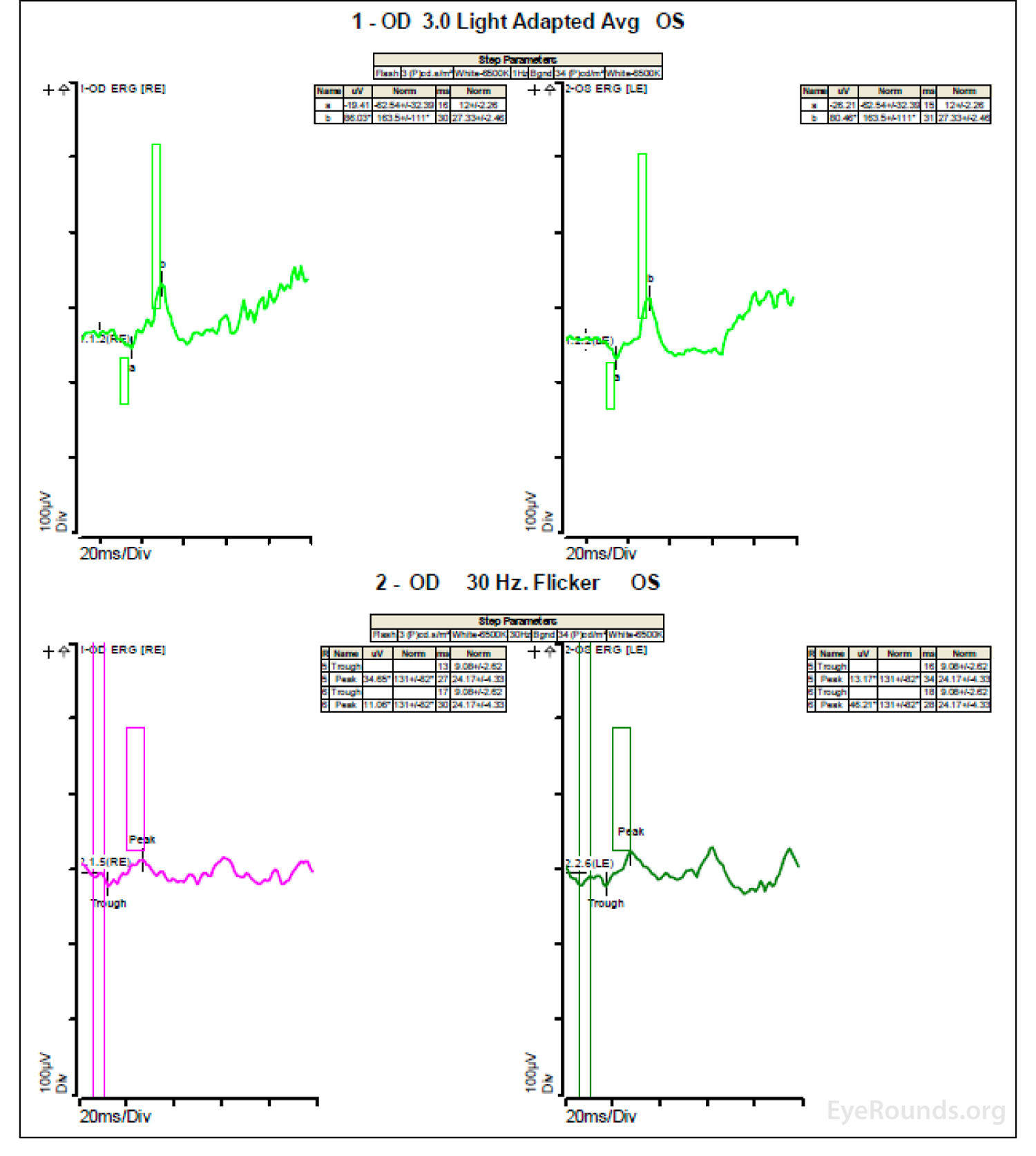 |
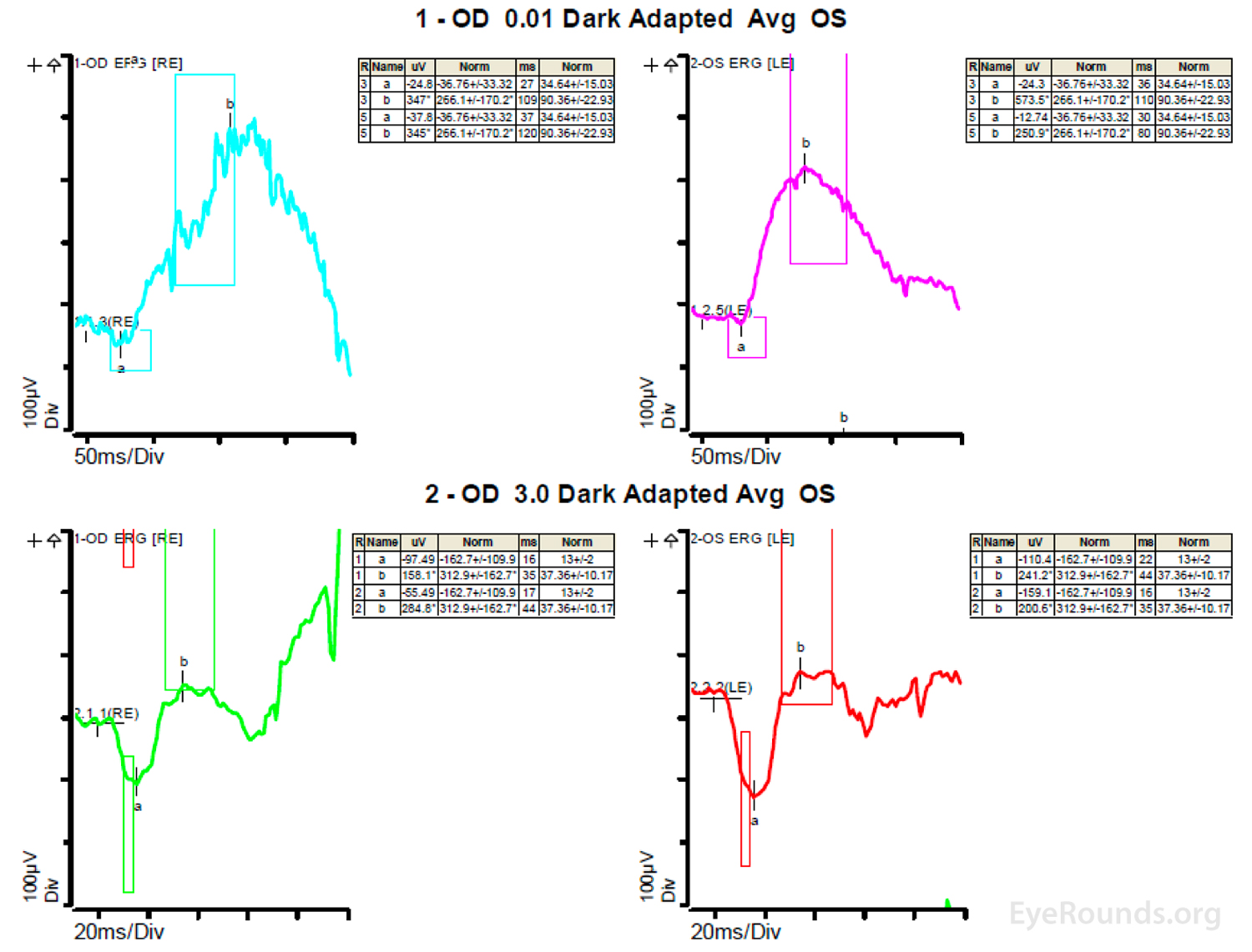
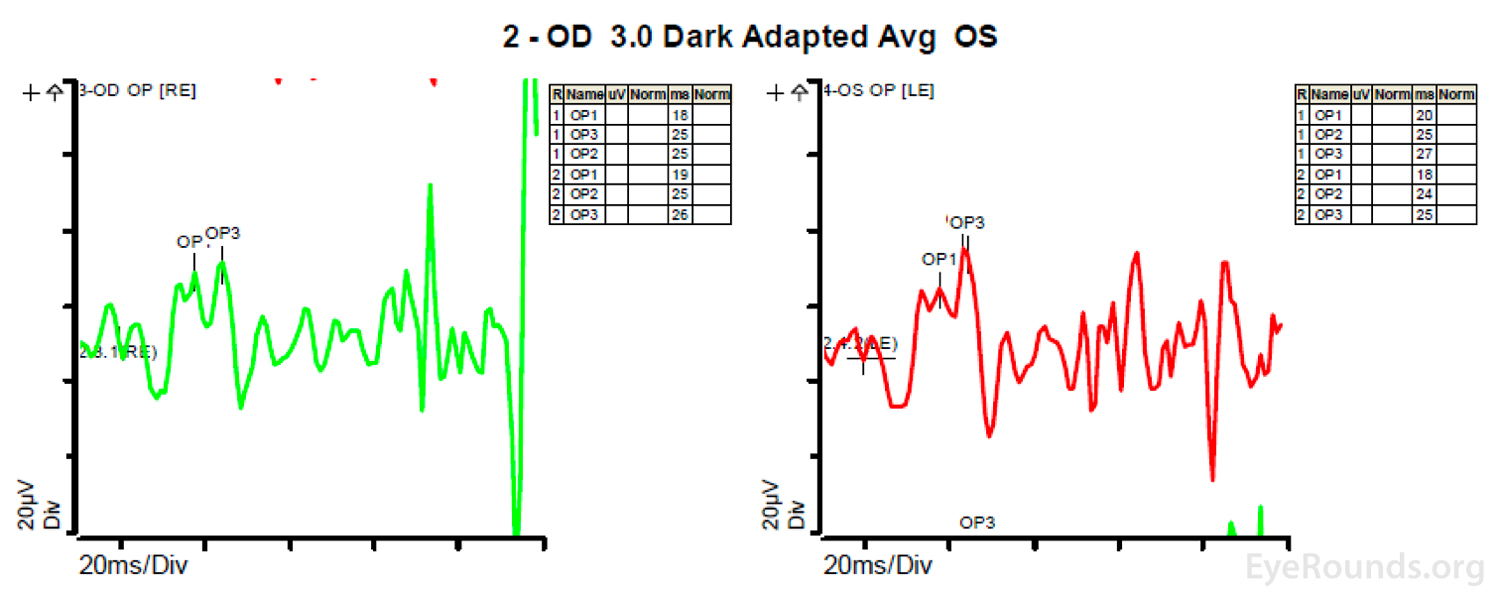
Figure 4: A, B, C. Figure 4A shows essentially normal dark adapted 0.01 cd.s/m2 rod responses and selective loss of the b-wave on 3.0 cd.s/m2 dark adapted responses. Figure 4B shows dark adapted 3.0 cd.s/m2 oscillatory potentials which are anomalous in configuration. Figure 4C shows low normal light adapted 3.0 cd.s/m2 responses and below normal 30 Hz flicker (cone isolated) responses OU.
Laboratory Values:
Homocysteine: 165.4 µmol/L (normal <= 10.0 µmol/L)
At this point in the patient's clinical course, there was concern for retinal changes due to homocystinuria, in the setting of significantly elevated blood homocysteine levels with increasing numbers of whitish intraretinal spots, both within the maculae and in the periphery. He was taking the recommended treatment of hydroxycobalalmin injections and betaine supplements, but his homocysteine level was still difficult to control. He had tried the recommended diet for his condition but had difficulty adhering to it. He was encouraged to work with a nutritionist and his other physicians in order to better control his homocysteine levels. He will return to the ophthalmology clinic for close follow up with repeat examination and imaging.
Homocystinuria, or accumulation of homocysteine in the blood and urine, is due to an inborn error in methionine metabolism that is commonly inherited in an autosomal recessive fashion. The prevalence of all types of homocystinuria is estimated to be 1: 344,000 [1]. There are different types of homocystinuria with the most common type caused by a deficiency in the enzyme cystathionine beta-synthase, which occurs in about 1:50,000 births [2 ,3]. This type of homocystinuria is usually detected on newborn screening tests.
To better describe the pathophysiology of homocystinuria, it is separated into three major categories based on the site of the metabolic defect. Type 1 is due to a deficiency in cystathionine beta-synthase, which converts homocysteine to cystathionine. Type 2 is due to defective methylcobalamin synthesis, which helps to convert homocysteine to methionine. Type 3 is due to an abnormality in methylene tetrahydrofolate reductase, which also helps to convert homocysteine to methionine [2]. These types have also been organized into broader categories: homocystinuria due to a defect in the transsulfuration pathway of homocysteine (type 1) and homocystinuria due to a defect the methylation pathway of homocysteine (types 2 and 3) [1]. Figures 5 and 6 show these different pathways and the sites of the enzymatic defects [4 ,5].
In this discussion, we will focus on homocystinuria due to methionine synthase deficiency, which is a defect in the methylation pathway of homocysteine. This specific deficiency is characterized by homocystinuria, hyperhomocysteinemia, and hypomethioninemia, without methylmalonic aciduria [6]. Methionine synthase catalyzes the methylation of homocysteine to methionine and can be caused by deficiency in methylene tetrahydrofolate reductase or by deficiency of methylcobalamin cofactors [4]. The patient in this case possesses the cb1G defect, or cobalamin G deficiency, leading to a deficiency in methylcobalamin cofactors [4]. Figure 6 demonstrates the location of the Cb1G defect [4 ,5].
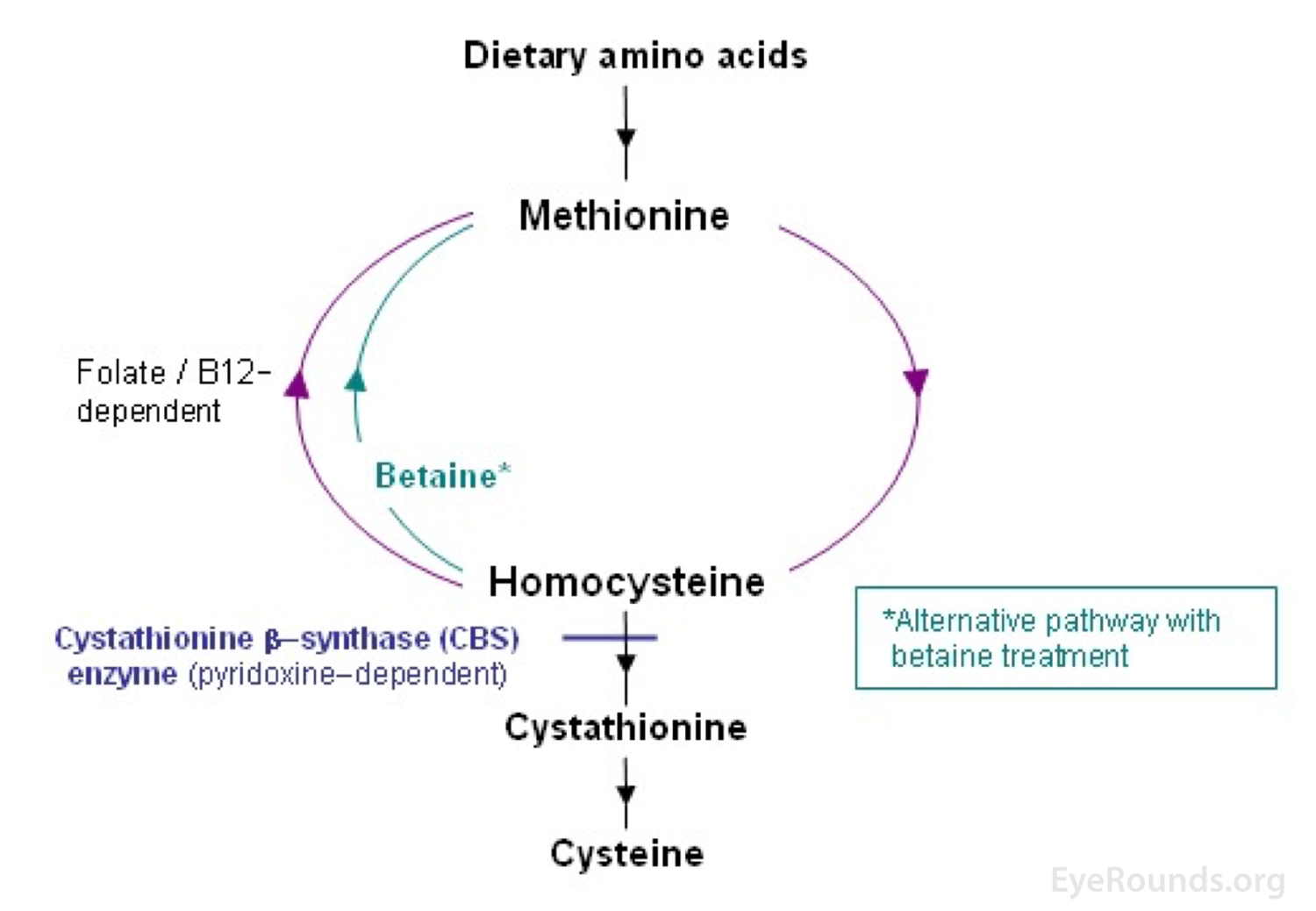
Figure 5. The metabolism of methionine to homocysteine and homocysteine to cysteine is shown. Specifically, the cystathionine B-synthase defect that leads to the most common form of homocystinuria is highlighted. This figure also demonstrates how betaine supplements help in homocystinuria. Taken from Sacharow SJ, Picker JD, Levy HL. Homocystinuria Caused by Cystathionine Beta-Synthase Deficiency. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, et al., editors. GeneReviews((R)). Seattle (WA); 1993.
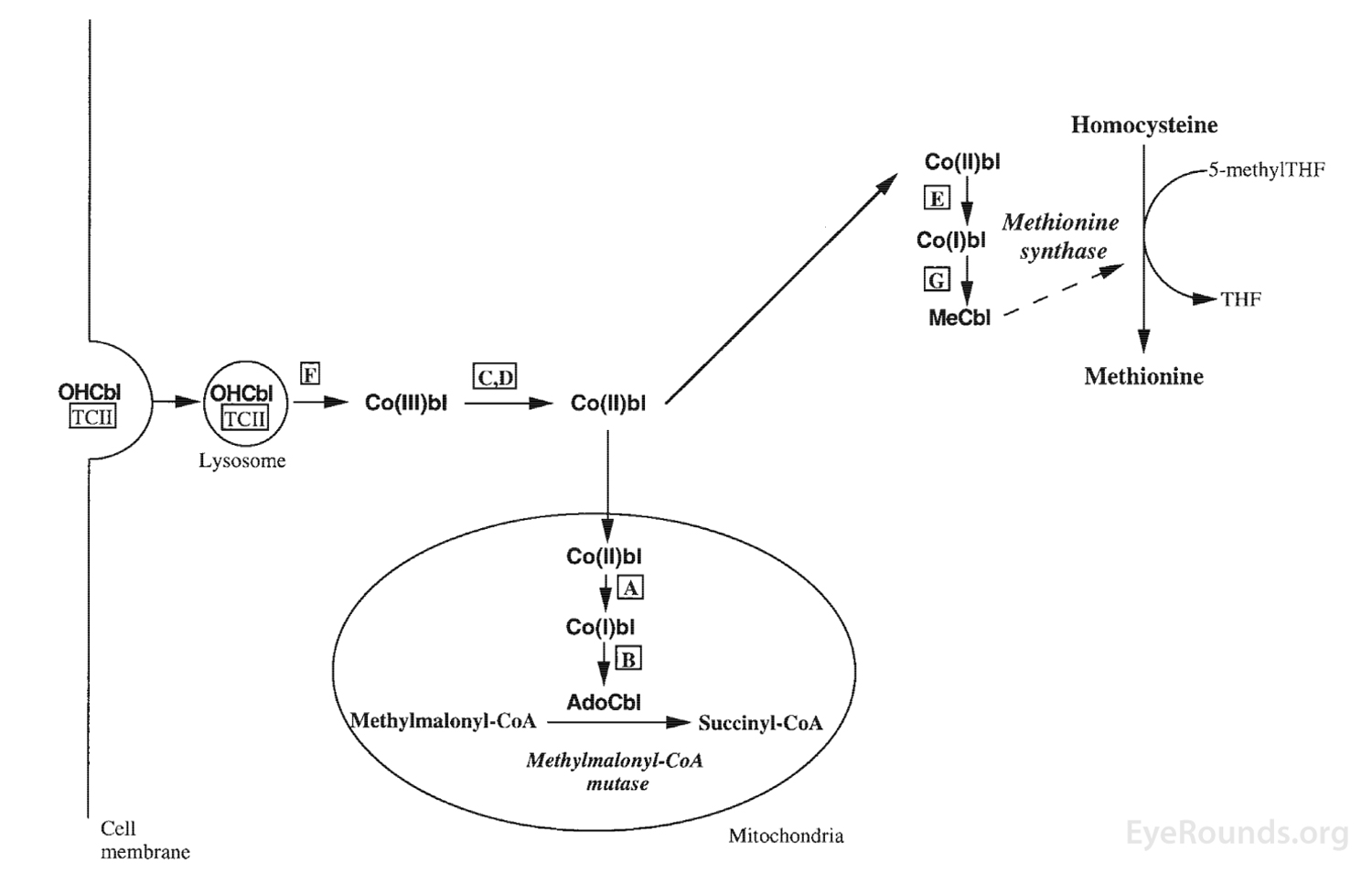
Figure 6. The production of methionine from homocysteine is shown, a process that requires hydroxycobalamin (OHCbI, Vitamin B12). The letters in this figure show the seven inborn errors of intracellular cobalamin metabolism. The patient in this case has the Cb1G defect, which leads to failure of methylation of the cobalamin cofactor needed for methionine synthase. Taken from Harding CO, Arnold G, Barness LA, Wolff JA, Rosenblatt DS. Functional methionine synthase deficiency due to cblG disorder: a report of two patients and a review. Am J Med Genet 1997;71(4):384-390. [PMID 9286442].
There have been few studies that offer theories for the pathophysiologic mechanisms underlying the ocular sequelae of homocystinuria. Poloschek et al. investigated ERG findings in those with methionine synthase deficiency and proposed that increased homocysteine levels decrease the transport of folates to photoreceptors, which in turn decrease intracellular anabolic processes, impacting cone photoreceptor function [7]. They also commented that hyperhomocysteinemia may lead to microvascular damage in the retina, leading to reduced oscillatory potentials. It may also have toxic effects on the neurons of the ganglion cell layer, leading to deformed flash visual evoked potential (VEP) responses [7]. Bacci et al. investigated OCT changes in patients with cobalamin C deficiency (cb1C) and commented that there may be reduced reuptake of methionine into the cone cell membrane during phototransduction, leading to a progressive maculopathy [8].
Ectopia lentis is prevalent in patients with homocystinuria. This is thought to be due to broken and matted zonular fibers that tend to break centrally and curl up against the lens [2]. The lens often subluxates inferiorly and anteriorly and can cause pupillary block [2 ,3]. In cystathionine beta-synthase deficiency, ectopia lentis presents in most patients by age 5 years old if the homocystinuria is left untreated [2 ,3].
Many patients with homocystinuria also develop nystagmus early on in life. [6-9]. In patients with cobalamin C deficiency (cb1C), nystagmus is present in 64% [10].
In patients with homocystinuria due to an error in cobalamin processing, specifically cobalamin C type deficiency (cb1C), there have been many reports describing the presence of a progressive maculopathy [8 ,9]. Of note, cobalamin C type deficiency is different than cobalamin G type deficiency due to the presence of methylmalonic aciduria. One study of patients with cb1C disease showed that macular changes were present in 72% of patients that underwent ocular examination [10]. There are various descriptions of the appearance of this maculopathy including pigment epithelial mottling, atrophic lesions, bull's eye appearance, and peripheral salt-and-pepper pigment changes [8 ,9].
Other ocular signs include progressive myopia, strabismus, iridodonesis, cataract, iris atrophy, retinal detachment, central retinal artery occlusion, optic atrophy, anterior staphylomas, sluggish pupillary response, and eyelid clonus [2 ,7 ,9 ,10].
There are many systemic signs and symptoms that are manifest with homocystinuria, such as developmental delay, failure to thrive, dental abnormalities, arachnodactyly, thromboembolic events, seizures, megaloblastic anemia, hypotonia, and feeding problems [3 ,4 ,6 ,9]. Thrombotic complications are obviously of high concern but have been more commonly reported in those patients with cystathionine beta-synthase deficiency rather than the other variants of homocystinuria [4].
OCT changes in patients with cb1C disease and maculopathy show loss of the external limiting membrane, myoid, ellipsoid, and interdigitation zones with preservation of the RPE in the foveal area [8]. In some patients, there are hyperreflective material in the parafoveal area just anterior to the RPE [8]. In patients with progressive maculopathy in cb1C disease, there is thinning and loss of the ganglion cell layer, outer nuclear layer, and photoreceptor layer on OCT [10 ,11].
ERGs have been performed in patients with homocystinuria, and evidence shows that these patients may show amplitudes that are reduced early in the course of the disease; often with the cones more affected than the rods [9]. In patients with methionine synthase deficiency, single flash cone b wave, flicker, and oscillatory potential responses were reduced below the fifth percentile of age-matched normals [7]. ERG under anesthesia on patients with cb1C disease shows 50% reduction in the scotopic b-wave and delay of the photopic a-wave and scotopic b-wave consistent with diffuse photoreceptor and bipolar cell dysfunction [8].
Homocystinuria is treated with vitamin supplementation and dietary restrictions. For patients with the cb1G defect, like the patient in this case, parenteral cobalamin supplementation is indicated as well as betaine supplementation, which acts as a methyl donor in the conversion of homocysteine to methionine (see Figure 5) [4 ,5]. Dietary restriction of protein is also indicated for some types of homocystinuria because it reduces methionine consumption which should in turn decrease homocysteine production [4 ,5]. Because methionine synthase deficiency may prevent the production of tetrahydrofolate, which can lead to folate deficiency and megaloblastic anemia, folate supplements can also be beneficial [4]. A study specifically looking at patients with maculopathy due to homocystinuria caused by cb1C disease noted that clinical progression of retinopathy is not prevented by early systemic therapy to reduce toxic metabolites [8].
Maculopathy (bilateral):
Ectopia Lentis:
EPIDEMIOLOGY OR ETIOLOGY
|
SIGNS/SYMPTOMS
|
TREATMENT/MANAGEMENT
|
|
Stiff HA, Drack AV. Homocystinuria. EyeRounds.org. posted May 30, 2018; Available from https://EyeRounds.org/cases/269-Homocystinuria.htm

Ophthalmic Atlas Images by EyeRounds.org, The University of Iowa are licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License.

Address
University of IowaLegal
Related Links
