
A 19-year-old female with a history of amblyopia in the right eye (OD) due to persistent fetal vasculature (PFV) presented to the University of Iowa Hospitals and Clinics (UIHC), Department of Ophthalmology to re-establish care. Her vision was stable. She denied active issues.
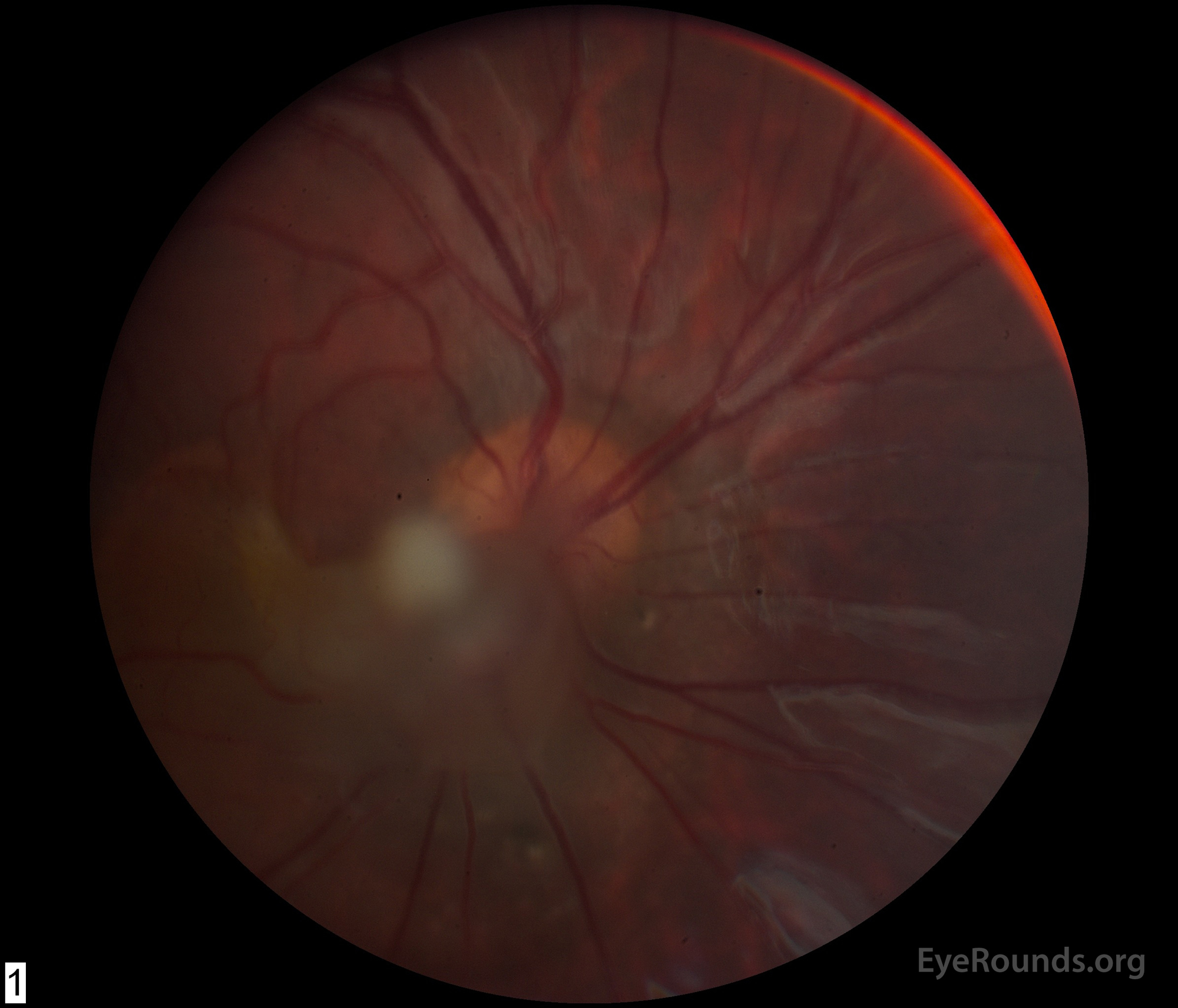
She was first seen at UIHC around 12 months of age and was diagnosed with amblyopia and esotropia OD secondary to PFV in this eye. Occlusion therapy was initiated (6 hours per day) and visual acuity improved. At 4 years of age, she underwent right lateral rectus resection resulting in improved cosmesis and a small amount of consecutive exotropia. By age 5, her vision was 20/60 and patching was tapered. She returned at age 6 with decreased visual acuity OD due to retinal traction by the PFV fibrovascular stalk (Figure 1A). She was then referred for pars plana vitrectomy OD and was subsequently followed locally.
No known allergies
Strabismus in two cousins and maternal grandfather
Non-contributory
Unremarkable except for what is mentioned in the history of present illness
Distance Visual Acuity, with correction (Snellen)
| Manifest Refraction | ||||
| Sphere | Cylinder | Axis | Distance VA | |
| Right | -6.75 | +1.00 | 091 | 20/80-1+1 |
| Left | -5.50 | +2.75 | 086 | 20/15-1 |
| Distance VA with both eyes: 20/15 | ||||
Right exotropia in primary gaze
Normal OU
 |
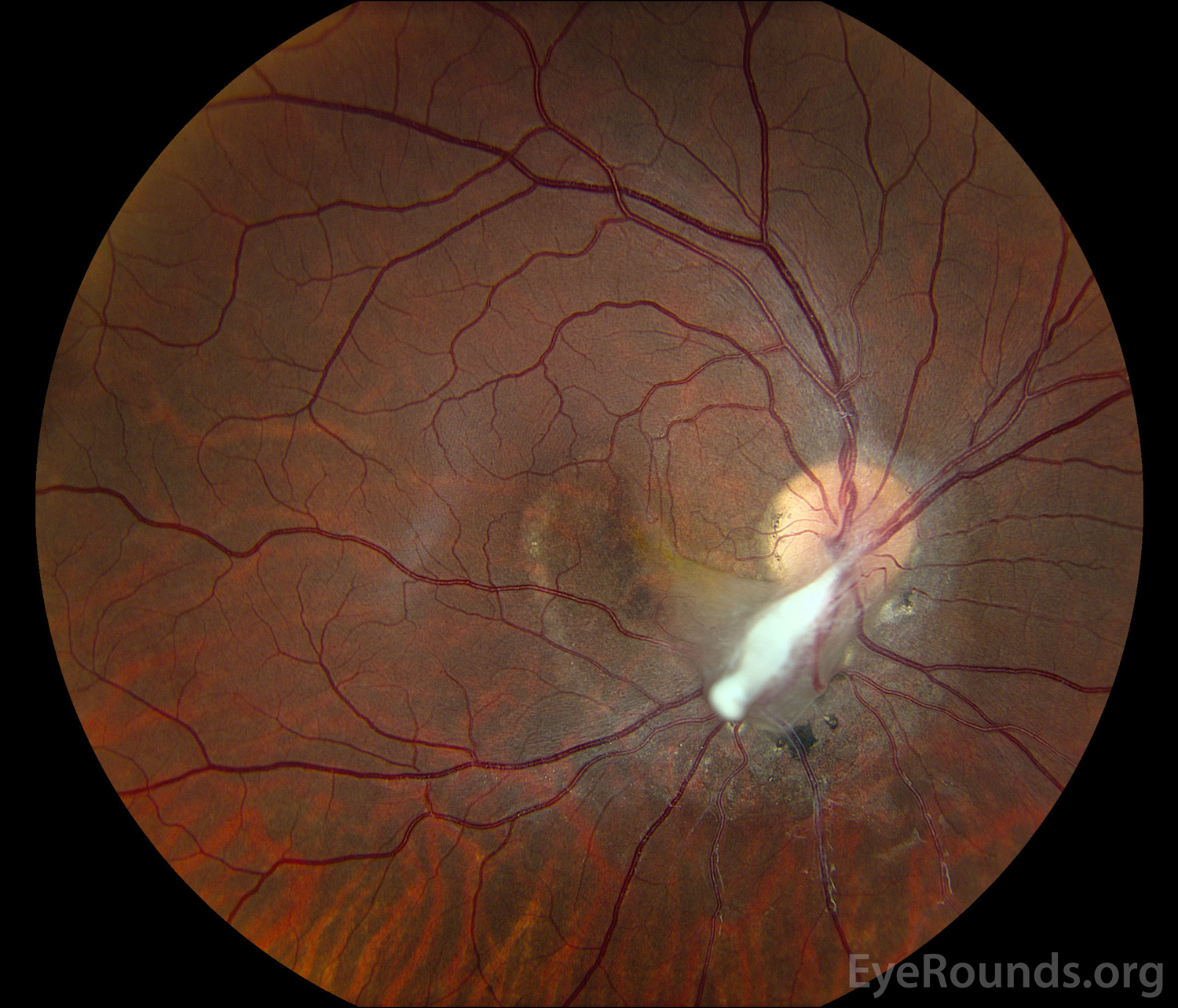 |
Figure 1: (A) Color fundus photograph OD at age 6 demonstrating a hazy fibrotic vitreous stalk projecting from the optic disc, which appears hyperemic with blurred margins. There is evidence of peripapillary and macular retinal traction and subretinal fluid based on fine retinal folds and loss of visible choroidal detail underlying the stalk. (B) Color Fundus Photograph OD at age 19 status post pars plana vitrectomy demonstrating the amputated stump of the white fibrovascular stalk with resolution of retinal traction. In the area of prior subretinal fluid, there is pigmentary change and clumping, forming what looks like a "watermark."
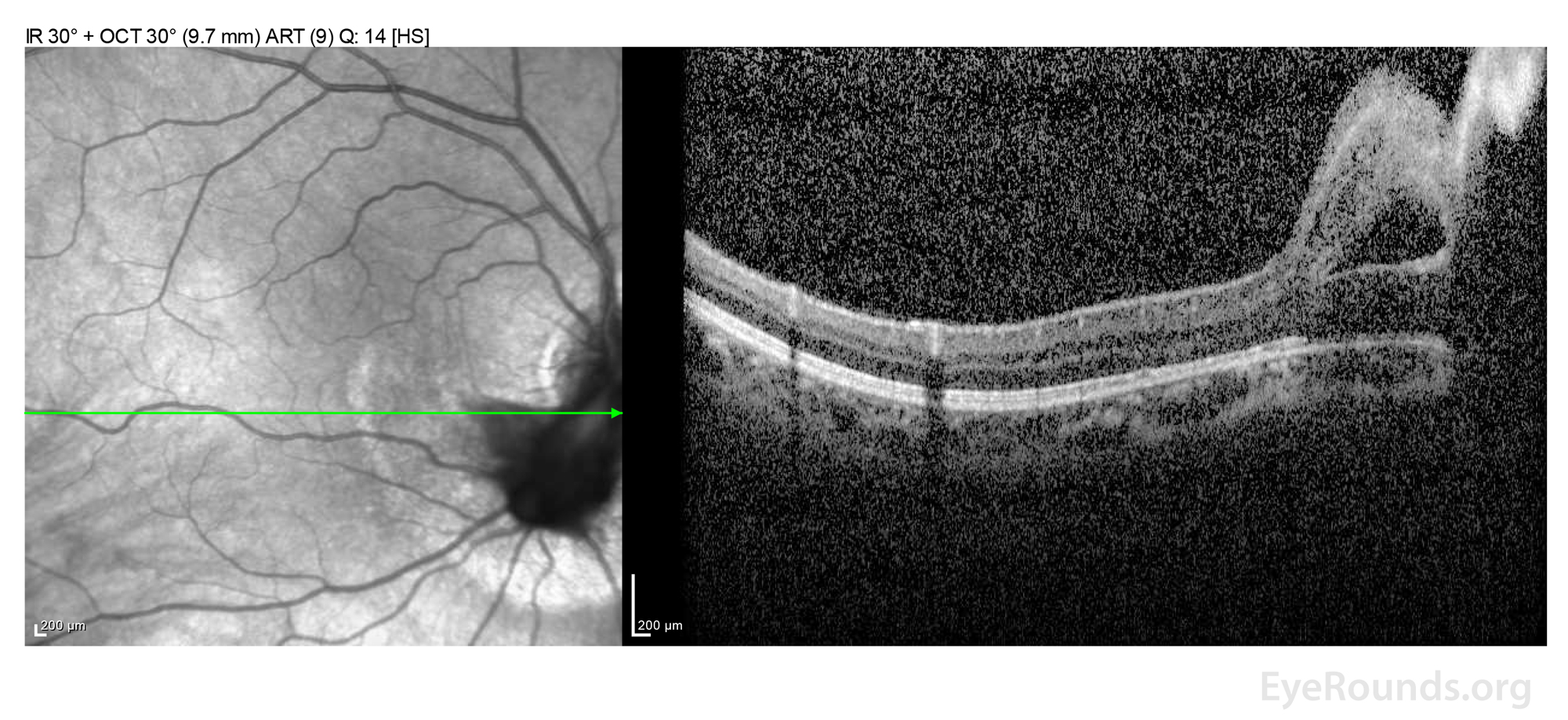
Figure 2: (A) OCT macula at age 19 demonstrating residual schisis of the peripapillary retina just inferior to the optic nerve. The en face infrared (IR) image on the left demonstrates the "water mark" where the peripapillary subretinal fluid resolved following pars plana vitrectomy with release of traction. There was no evidence of persistent subretinal fluid.
Persistent fetal vasculature
Most cases of persistent fetal vasculature (PFV), previously known as persistent hyperplastic primary vitreous (PHPV), are sporadic and non-heritable congenital eye disorders. Nevertheless, some cases of autosomal dominant and autosomal recessive inheritance have been recorded[1] . Although no set of genes can account for the majority of PFV, the ATOH7 and NDP genes have been implicated in autosomal recessive and dominant cases, respectively [2 ,3] . PFV is generally diagnosed soon after birth and typically presents unilaterally in normal full-term infants without associated systemic findings [4] . However, rare bilateral lesions may be associated with trisomy 13 [5] .
The developing lens of the fetus is fed by a network of vessels called the tunica vasculosa lentis. This network surrounds the posterior and lateral lens and comes from the hyaloid artery, which originates from the ophthalmic artery. The regression of this fetal vascular system starts in the third month of gestation and ends in the eighth month[6] . Some models suggest that this fetal vascular system regresses through apoptosis and macrophage activation to allow uninhibited transmission of light through the lens and to the retina. Ocular astrocytes appear to play a vital role in this process through precise migration and by facilitating key signaling pathways. Studies speculate that impaired endolysosomal signaling from mutation of the Crybal gene and impaired migration from increased Aquaporin 4 expression in astrocytes may prevent activation of the necessary macrophage-mediated involution, thus leading to PFV [7 ,8] . Another proposed model details unrepressed vascular endothelial growth factor (VEGF) in the hyaloid vascular system due to loss of the Arf tumor-suppressor gene [9 ,10] . Nevertheless, the exact mechanism of PFV formation is still largely unknown.
PFV is divided into three subcategories: anterior, posterior, or a combination of the two. In anterior PFV, remnants of the tunica vasculosa lentis fail to regress, leading to a retrolental fibrovascular membrane that can obstruct the visual axis and cause early cataract formation. Elongated ciliary body processes are commonly seen in anterior PFV. In posterior PFV, failed degradation of the hyaloid artery results in a fibrovascular stalk connected to the vitreous and retina. The stalk may pull on surrounding structures causing retinal folds, a tractional retinal detachment, retinal or vitreous hemorrhages, retinal dysplasia, or hypoplasia of the optic nerve and macula. Microphthalmia is associated with both anterior and posterior types of PFV [4 ,11] and is often one of the first signs of PFV in an infant, along with cataract and leukocoria [12] . As time passes, the eye affected by PFV may develop glaucoma from angle-closure and phthisis bulbi. Enucleation may be required if PFV is not treated at an early stage [13] .
Given the presence of leukocoria, retinoblastoma must be on the differential diagnosis for PFV although it is rarely associated with microphthalmia and should show a distinct mass on imaging. PFV can be differentiated from other causes of tractional detachment such as familial exudative vitreoretinopathy (FEVR), Norrie disease, and incontinentia pigmenti by its unilateral presentation as opposed to these bilateral hereditary conditions [14] . Coats disease should also be included on the differential as it may present with leukocoria, retinal detachment, and/or strabismus.
Direct visualization via ophthalmoscopy can be difficult due to frequently opaque ocular media. If leukocoria is not severe, visualization of the hyaloid fetal vasculature provides the best indicator of PFV. A fibrovascular stalk may be seen posteriorly connecting to the retina with associated retinal folding and hemorrhages, while elongated ciliary processes and elongated iris vessels may be present anteriorly [13] .
The resulting leukocoria and/or retinal damage may cause decreased visual acuity in the affected eye, an RAPD, strabismus, or eventual amblyopia. The anterior chamber depth should also be evaluated due to the risk of glaucoma [15] .
PFV can be visualized with several imaging modalities such as fluorescein angiography, fundus photography, computed tomography (CT), and magnetic resonance imaging (MRI). Fluorescein angiography may reveal a persisting anterior tunica vasculosa lentis extending to the iris margin. If leukocoria is not severe, fundus photography may visualize a tortuous fibrotic vascular remnant emanating from the optic nerve, known as a Bergmeister papilla, a fibrovascular stalk, or retinal folding, tenting, or detachment. If media opacity obscures visualization of the posterior pole, ocular ultrasound, CT, and MRI may provide useful information. Retinal detachment and the fibrovascular hyaloid remnant may also be seen with ultrasonography [16] . CT can be used to detect microphthalmia, the lack of calcification, increased vitreous chamber density, enhancement of abnormal intravitreal tissue, a shallow anterior chamber, or a small, irregular lens. MRI findings may include a flattened lens, a shallow anterior chamber, or enhancement of anterior segment structures in the anterior type of PFV. A hyperintensity of the vitreous chamber on T1-weighted images can be seen in combined anterior-posterior or isolated posterior PFV types. Both CT and MRI may show enhancement of abnormal intravitreal tissue, a linear structure suggestive of the hyaloid canal, or an enhancing retrolental mass [13] .
A wide spectrum of PFV severity translates into a similarly wide range of treatment options including observation, amblyopia therapy, and surgery.
Surgery to remove the fibrovascular remnants is recommended to prevent the development of retinal detachments, glaucoma, and phthisis. Vitrectomy is recommended in the case of a significantly deformed retina, severe intravitreal hemorrhage, progressive anterior chamber shallowing, or retinal detachment. A plaque on the posterior lens can be removed to increase visual acuity, and the stalk can be amputated to allow for continued eye growth. On the other hand, surgery is contraindicated in cases with no light perception vision, unrecordable visual evoked potentials, or severe RAPD.
In anterior PFV, lensectomy can be performed to remove the cataract and a vitreous cutter may be used to separate the fibrovascular retrolental membrane from the ciliary processes. In posterior PFV, vitrectomy allows for hyaloid stalk amputation and membrane peel may be needed to release retinal traction and to repair any retinal detachment. Amblyopia therapy should follow any surgical intervention [4 ,14] .
ETIOLOGY
|
SIGNS
|
SYMPTOMS
|
TREATMENT/MANAGEMENT
|
Mai A, Provencher LM, Oetting TA. Persistent Fetal Vasculature. EyeRounds.org. posted May 31, 2018; Available from https://EyeRounds.org/cases/270-persistent-fetal-vasculature.htm

Ophthalmic Atlas Images by EyeRounds.org, The University of Iowa are licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License.

Address
University of IowaLegal
Related Links
