
Severe left eye pain
A 78-year-old Caucasian woman presented with a two-week history of severe left eye pain and redness. The pain was initially described as periorbital "fullness," and she was prescribed oral antibiotics by an outside provider for a presumed sinus infection. The pain gradually worsened to a "sharp" and "stabbing" pain, and she was prescribed topical antibiotics and artificial tears for a presumed conjunctival abrasion. Despite treatment, her left eye pain and redness continued to worsen such that her discomfort awakened her during the night, which made sleeping difficult. On presentation, she also reported photophobia, blurry vision, and a foreign body sensation in her left eye (OS). She denied any symptoms in her right eye. She reported a history of intermittent red rashes on her legs. The remainder of her review of symptoms was negative.
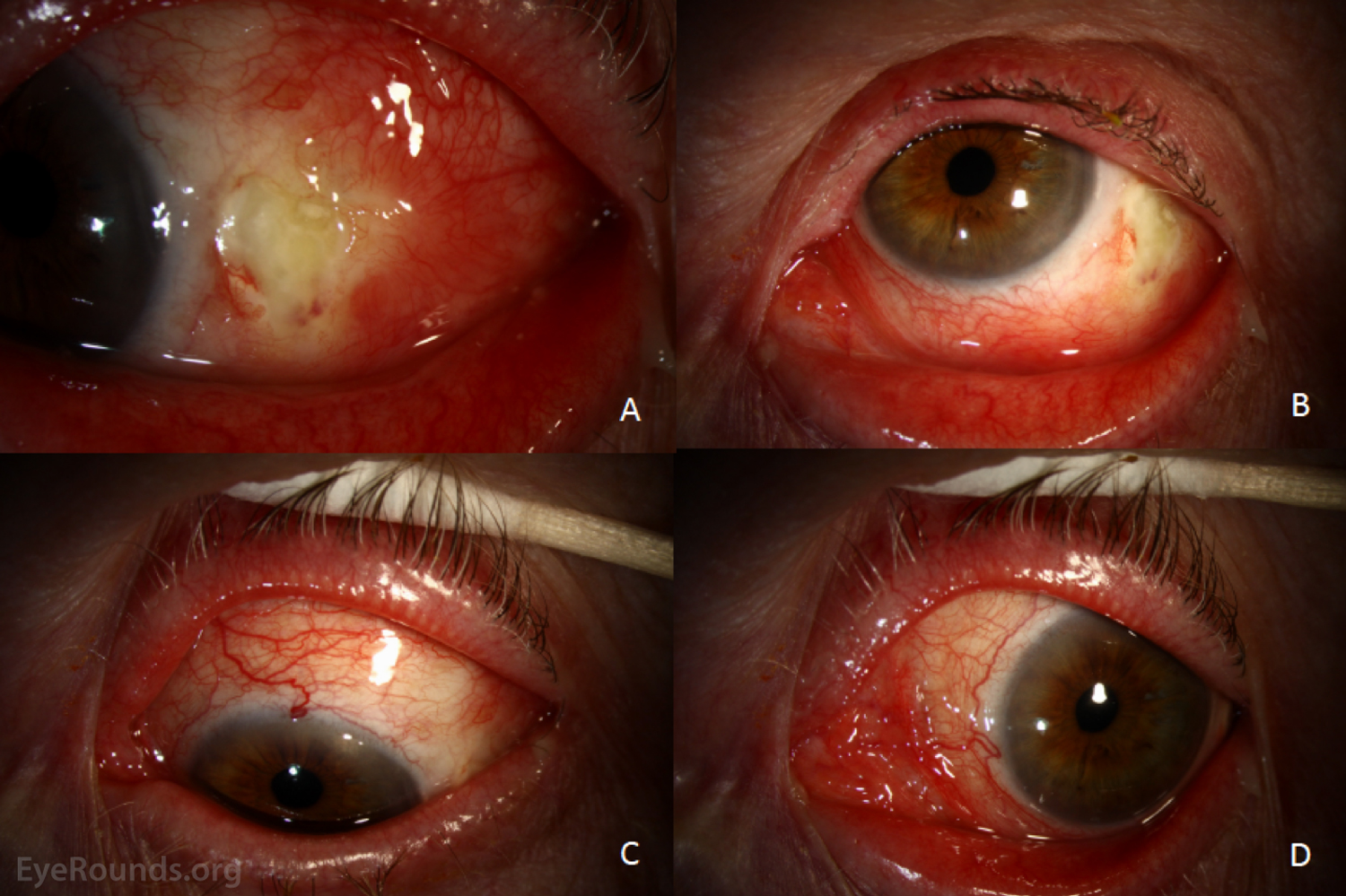
Figure 1: External photographs at initial presentation. The left eye is shown in right gaze (A), up gaze (B), down gaze (C), and left gaze (D). There is diffuse injection following the instillation of phenylephrine 2.5%. There is a conjunctival epithelial defect at the temporal limbus anterior to the lateral rectus insertion
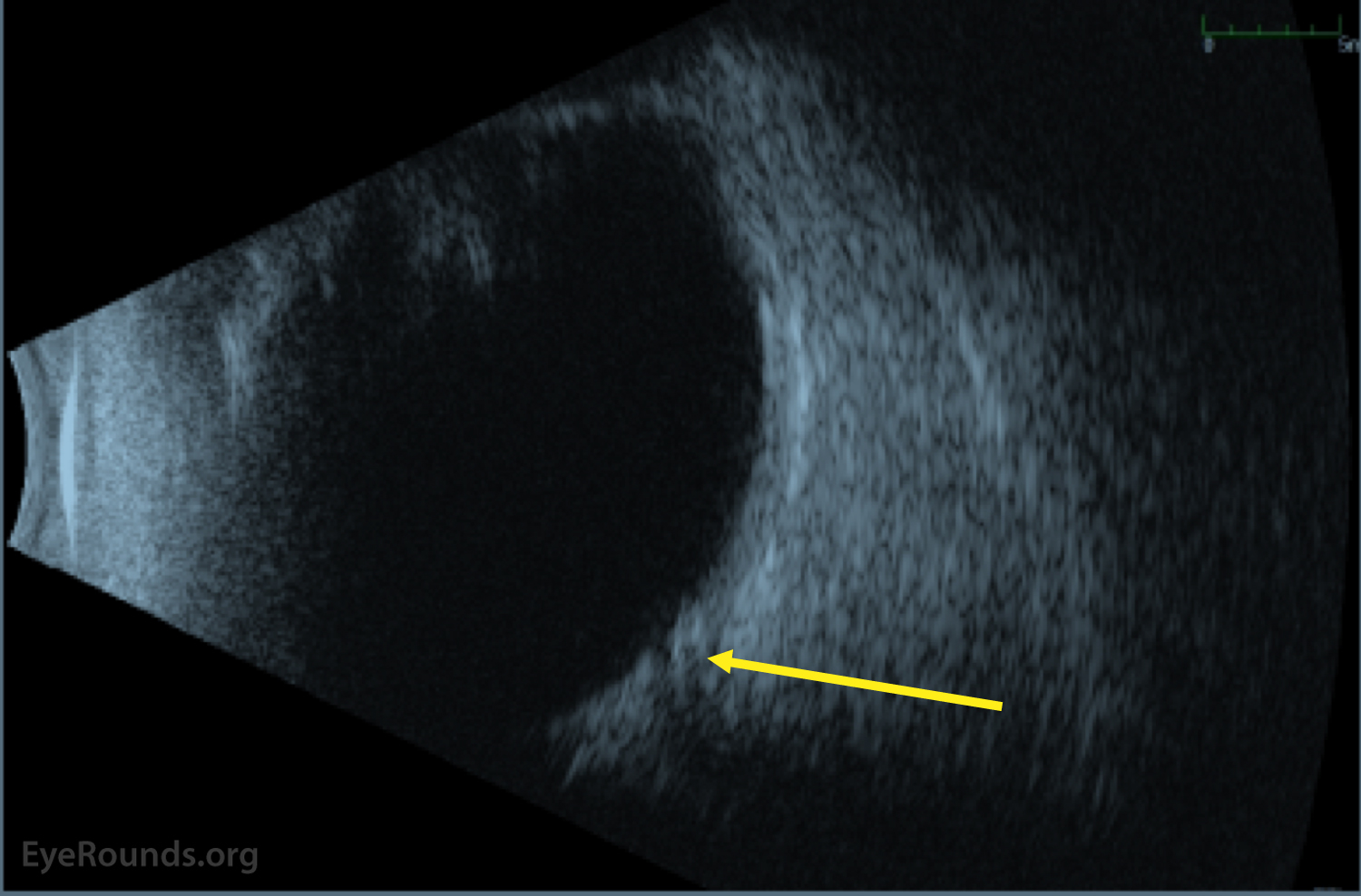
Figure 2: B-scan ocular ultrasound upon initial presentation. There is no evidence scleritis or vitreous inflammation. There is however evidence for early cupping of the optic nerve (yellow arrow).
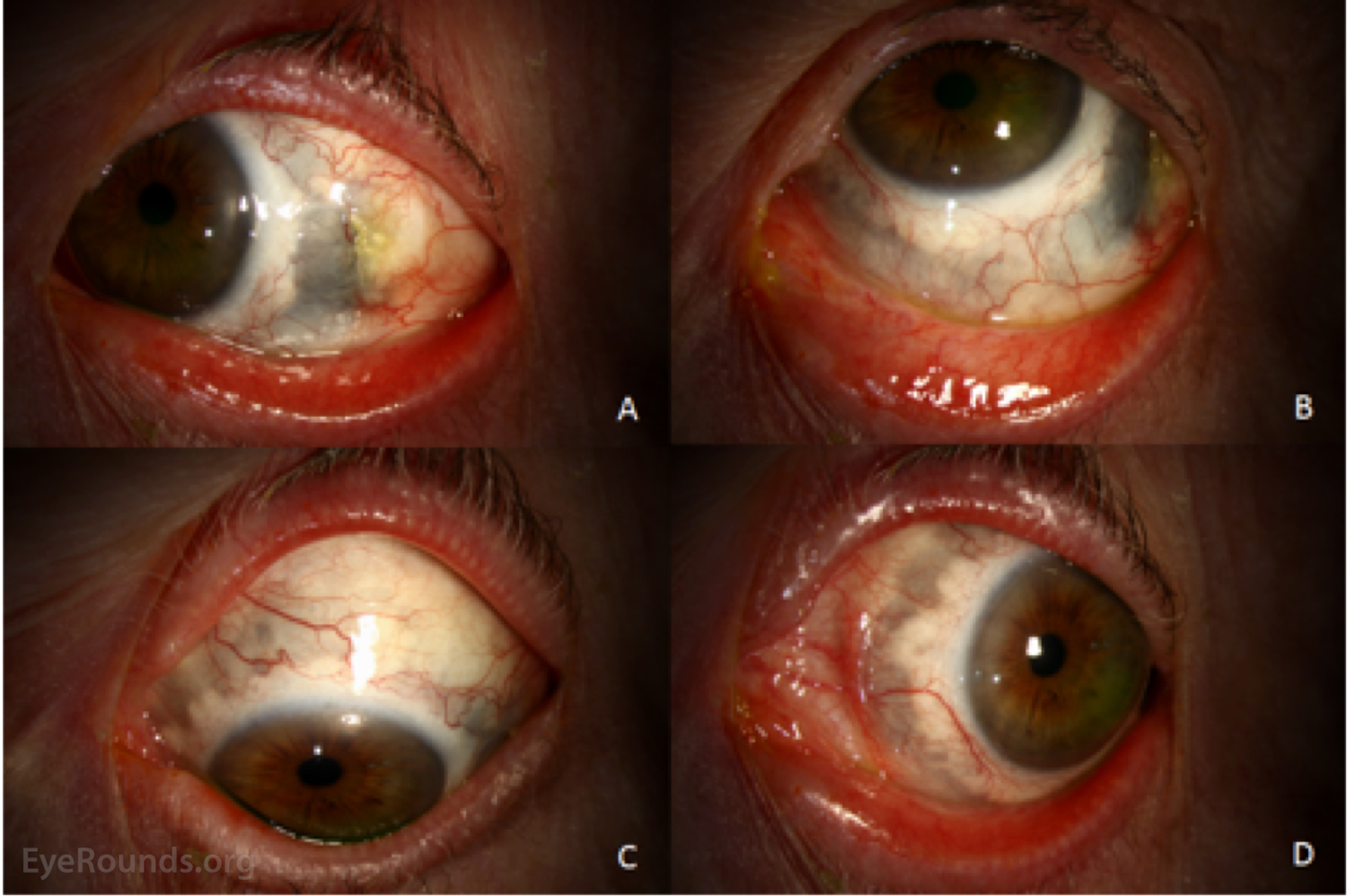
Figure 3: External photographs at latest follow-up. The left eye is shown in right gaze (A), up gaze (B), down gaze (C), and left gaze (D). The conjunctival injection is markedly improved. The temporal conjunctival defect is beginning to epithelialize. Brown or dark pigmentation is now present from approximately 1 o'clock to 11 o'clock (clockwise). This represents direct visualization of the choroid and ciliary body due to loss of scleral tissue from the inflammatory process.
The sclera is an opaque outer coating of the eye that extends from the cornea to the optic nerve. Posteriorly, the outer layers are continuous with the dura of the optic nerve and the inner layers are continuous with the lamina cribrosa. This structure maintains the shape of the globe and serves as the attachment site for extraocular muscles. It is thinnest behind the insertion sites of the rectus muscles and thickest at the posterior pole.
Three vascular plexi supply the outer layers of the eye and include the conjunctival plexus, superficial episceleral plexus, and the deep vascular plexus. The conjunctival plexus lies within the conjunctiva, has no particular pattern, is freely mobile, and appears bright red when inflamed. The superficial episcleral plexus lies within the superficial episclera, has a radial configuration, is mobile over deeper layers, and appears salmon pink when inflamed. The deep vascular plexus lies deep to tenon's capsule and directly over the scleral stroma, has a criss-cross pattern, is immobile, and appears violaceous when inflamed.
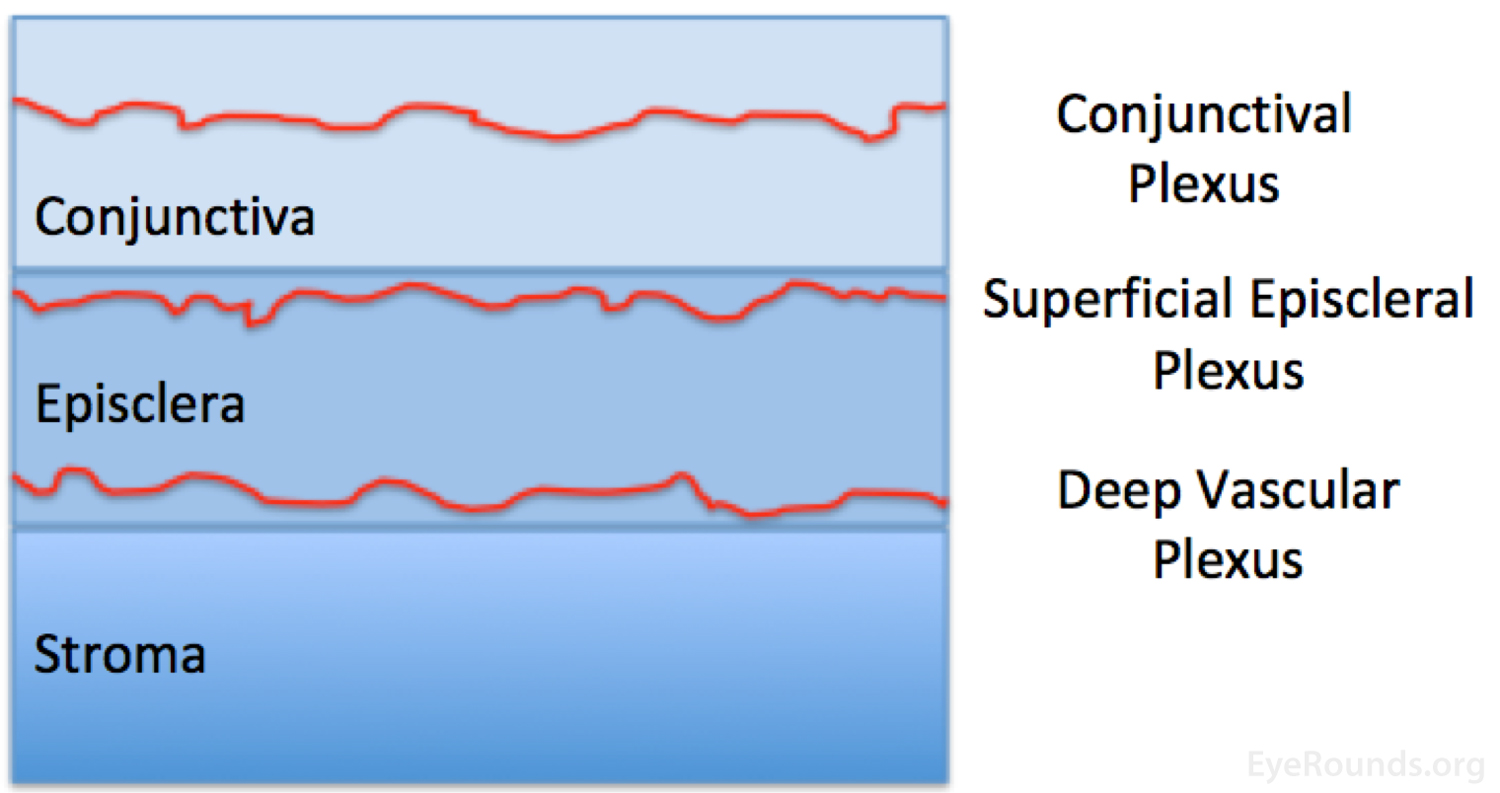
Figure 4: Anatomical depiction of the conjunctiva, episclera, and scleral stroma, and the approximate location of the conjunctival, superficial episcleral, and deep vascular plexi.
The sclera is composed of three layers. These layers include the episclera, stroma, and lamina fusca. The episclera is the outermost connective tissue layer and contains the superficial episcleral plexus and deep vascular plexus. The stroma is the primarily avascular middle layer and is composed of type I collagen. The lamina fusca is the inner layer, is composed of connective tissue, and anchors the sclera to the underlying choroidal tissue.
Scleritis is classified as either non-necrotizing or necrotizing scleritis. Non-necrotizing scleritis includes diffuse scleritis and nodular scleritis. Necrotizing scleritis includes necrotizing scleritis with inflammation and without inflammation. Scleritis may also be classified as anterior or posterior if inflammation is anterior or posterior to the insertion site of the extraocular muscles, respectively [1].
In general, scleritis is more common in women than men and usually occurs during the fifth decade of life [2]. The prevalence and incidence are 5.2 per 100,000 persons and 3.4 per 100,000 person-years, respectively [2]. The most common form is diffuse scleritis and the second most common form is nodular scleritis [1]. The least common form is necrotizing scleritis with inflammation [1]. In general, anterior scleritis is more common than posterior scleritis.
Although many cases of scleritis are idiopathic, this entity is often associated with an underlying autoimmune/inflammatory disorder. The most commonly implicated etiology is rheumatoid arthritis (RA), which is a chronic inflammatory polyarthritis that may involve multiple organ systems [1 ,3 ,4]. Other common autoimmune/inflammatory entities causing scleritis include granulomatosis with polyangitis (GPA), systemic lupus erythematous (SLE), ankylosing spondylitis (AS), polyarthritis nodosa (PAN), and giant cell arteritis (GCA) [1 ,3]. Infectious etiologies are less commonly implicated and include varicella zoster virus (VZV) and herpes simplex virus (HSV), pseudomonas, Lyme disease, tuberculosis, and syphilis [1 ,3 ,5]. In some cases, scleritis may be the only manifestation of an underlying systemic illness [6].
Scleritis is an immune-related inflammatory process of the sclera. Although autoimmune/inflammatory disorders and infectious pathogens are often associated with scleritis, this disorder is often idiopathic. Furthermore, pro-inflammatory antigens that initiate inflammation as well as the precise role of immune complexes have not been established[7]. It has been suggested that the avascularity of the scleral stroma results in accumulation of pro-inflammatory antigens, which trigger the development of scleritis [7]. The hallmark of scleral inflammation is stromal infiltration with T-cells and macrophages, which may lead to subsequent necrosis and scleral thinning in cases of necrotizing scleritis [7].
Typical ocular symptoms of diffuse anterior scleritis, nodular anterior scleritis, and necrotizing scleritis with inflammation are gradual onset eye pain and redness that may be unilateral or bilateral [1]. The eye pain is characterized as boring pain that is worse with eye movement, during the night, and involves adjacent structures of the face [1 ,7]. Uniquely, necrotizing anterior scleritis without inflammation (scleromalacia perforans) presents with minimal pain and redness despite significant vision loss due to acquired astigmatism in the setting of significant scleral thinning [1]. Posterior scleritis presents with eye pain as previously described as well as vision loss due to damage of the adjacent retina and optic nerve [1 ,8].
Typical ocular signs of anterior scleritis include an edematous sclera, globe tenderness, and prominent deep episcleral vessels [1]. As previously mentioned, these vessels have a crisscross pattern, do not blanch with administration of topical 2.5-10% phenylephrine, and are immobile with a cotton-tipped applicator [1]. In general, diffuse anterior scleritis presents with widespread scleral edema and erythema without necrosis. Nodular anterior scleritis presents with localized scleral edema and erythema with a nodular apperance. Necrotizing anterior scleritis with inflammation presents with scleral edema and erythema with a bluish discoloration of the involved sclera. Blue discoloration is the result of scleral thinning and visualization of underlying uvea. Necrotizing anterior scleritis without inflammation (scleromalacia perforans) presents with minimal scleral edema and erythema with a blue or yellow discoloration of the involved sclera. Yellow discoloration is the result of localized tissue infarctions. Posterior scleritis may present with thickening of the posterior sclera and choroidal effusions demonstrated on B-scan ultrasound.
Scleritis is a clinical diagnosis that involves a thorough ocular history and examination. However, systemic disease associated with scleritis is common [5]. Prior research suggests that around 28% of cases are related to systemic disease with RA accounting for 12.8% and systemic vasculitis accounting for 7.8% of cases [5]. Thus, consultation from a rheumatologist is prudent to aid in establishing an underlying diagnosis [9]. Systemic work-up includes a detailed history and physical examination focusing on the musculoskeletal, integumentary, and cardiopulmonary systems. Routine laboratory testing may include a complete blood count (CBC) with differential, complete metabolic panel (CMP), urinalysis (UA) and microscopy, erythrocyte sedimentation rate (ESR), C-reactive protein (CRP), rheumatoid factor (RF), anti-cyclic citrullinated peptide (anti-CCP), antinuclear antibody (ANA), antineutrophil cytoplasmic antibodies (ANCA), uric acid, angiotensin converting enzyme, and serologies for syphilis and lyme disease [1 ,9-16]. Routine imaging includes a chest radiograph to exclude pulmonary manifestations of systemic illness.
| Etiology | Diagnostic Criteria |
| Rheumatoid Arthritis (RA) | Joint pain and stiffness that involve the smaller joints first, especially in the hands and feet, with positive rheumatoid factor and anti-cyclic citrullinated peptide. |
| Systemic Lupus Erythematosus (SLE) | Requires four of the 11 criteria: Malar rash, discoid rash, photosensitivity, mucosal ulcers, arthritis (non-erosive), cardio-pulmonary involvement, neurologic disorder, renal disorder, hematologic disorder, antinuclear antibodies, and/or immunologic disorder. |
| Ankylosying Spondylitis (AS) | Joint pain and stiffness that involve the lower back, entheses that involves ligaments of the spine and the back of the heel, plain film of the pelvis revealing fusion of sacroiliac joints, and plain film of lumbar spine revealing bamboo sign. HLA-B27 is not among diagnostic criteria but does increase risk. |
| Granulomatosis with Polyangiitis (GPA) | Nephritic syndrome detected by urine tests, pulmonary vasculitis detected by chest X-ray, and tissue biopsy. |
| Polyarteritis Nodosa (PAN) | Requires documented vasculitis with three of the following: weight loss, livedo reticularis, testicular pain, myalgias, neuropathy, elevated diastolic blood pressure, elevated creatinine, hepatitis B virus infection, arteriographic abnormalities, biopsy of small or medium sized arteries with polymorphonuclear cells. |
| Giant Cell Arteritis (GCA) | Requires three of the following: age > 50, ESR > 50, temporal headaches, scalp tenderness, biopsy proven GCA. Other findings include: constitutional symptoms, jaw claudication, vision loss, and a history of polymyalgia rheumatica (PMR). |
Figure 5: Diagnostic criteria for the most common underlying systemic diseases causing scleritis including RA, SLE, AS, GPA, PAN, and GCA.
Our patient did not meet diagnostic criteria for the autoimmune/inflammatory or infections described above. However, our patient had a previous diagnosis of leukocytoclastic vasculitis. This entity is diagnosed via punch biopsy of cutaneous lesions, which reveals a neutrophilic small vessel vasculitis within the dermis [17]. In short, the clinical presentation of leukocytoclastic vasculitis includes palpable monomorphous purpura [17]. Lesions may also present as urticarial plaques. Dependent areas of the body such as the lower extremities and buttocks are usually involved and cutaneous findings are usually symmetric across the body[17]. Extracutaneous involvement occurs in nearly 30% of patients[17]. Although leukocytoclastic vasculitis is often idiopathic, an extensive work-up is indicated to rule out an underlying cause.
Treatment of scleritis aims to halt inflammation in order to reduce scleral thinning and subsequent damage to ocular structures. As illustrated in this case, treatment may involve multiple agents depending on the severity of disease and aggressive therapy is often required before stabilization of the disease can be obtained. In general, management options include non-steroidal anti-inflammatory drugs (NSAIDs), corticosteroids, and immunomodulatory agents.
Non-necrotizing scleritis is treated with topical corticosteroids, such as prednisolone acetate, with minimal risk of scleral thinning [18 ,19]. Systemic side effects of NSAIDs include gastrointestinal upset and interstitial nephritis. Recent research suggests that subconjunctival corticosteroid injections (SCIs) may also be considered [20]. A prior retrospective, non-comparative, interventional case series showed complete resolution of signs and symptoms within six weeks of subconjunctival corticosteroid injection in 36 of 38 patients with scleritis resistant to prior local or systemic therapy [20]. Furthermore adverse events were low without any cases of scleral melting or perforation [20].
Necrotizing scleritis and cases of non-necrotizing scleritis refractory to topical corticosteroids and NSAIDs are treated with oral corticosteroids, such as prednisone. Similar to this case, a reasonable starting dose is 1 mg/kg/day, which may be titrated up until inflammation is controlled [21]. In general, oral corticosteroids are continued for one month after scleritis is under control before being slowly tapered off. Our patient had steroids slowly tapered off over a seven-month interval. Systemic side effects include weight gain, mood changes, loss of bone density, insomnia, gastrointestinal upset, and hyperglycemia. The patient should be referred back to his/her primary care provider for consideration of bisphosphonates and monitoring of blood glucose levels.
Severe necrotizing scleritis and co-existing autoimmune disease are often treated with biologic agents. These agents may also be used as "corticosteroid-sparing" medications in patients with significant side effects to steroids. A reasonable starting dose for methotrexate is 15 mg weekly, which may be titrated up to 25 mg weekly [22]. Daily folic acid 1 mg is co-administered to prevent folic acid deficiency and subsequent megaloblastic anemia. Systemic side effects include gastrointestinal upset and transaminitis, which must be carefully monitored by the prescribing provider. Other immunomodulatory agents that may be considered include azathioprine, cyclophosphamide, mycophenolate mofetil, cyclosporine, and infliximab. In general, the most common indications for surgery include scleral and corneal thinning that is high risk for perforation or that is advanced to the point of perforation, cataract surgery, and glaucoma surgery [23]. Various graft tissues exist and include preserved donor sclera, fascia lata, periosteum, aortic tissue, and synthetic Gore-Tex [23-25]. Although temporarily helpful, surgery does not solve the underlying problem and the inflammation must be controlled to protect the graft and the patient's eye [23-25]. Corneal perforations may be treated conservatively with corneal glue until inflammation is under control, but a lamellar or perforating keratoplasty may be necessary [23].
The resolution of scleritis is highly variable and depends on the classification of the disease process as well as associated ocular complications and the presence of an underlying systemic process. In general, uncomplicated non-necrotizing anterior scleritis responds well to therapy and usually resolves within two months of appropriate therapy[19]. Cases of necrotizing scleritis can last much longer and inflammation may linger for many months prior to remission[1]. Furthermore, patients with mild to moderate scleritis without necrosis usually maintain good vision while patients with necrotizing scleritis are at higher risk for visual loss. Prior research suggests that roughly 37% of these patients ultimately lose two or more lines of visual acuity[26]. In patients with scleritis, 42% develop anterior uveitis, 14% develop peripheral ulcerative keratitis (PUK), 13% develop glaucoma, 17% develop cataract, and 6% develop fundus abnormalities such as retinal or choroidal detachments [26].
EPIDEMIOLOGY OR ETIOLOGY
|
SIGNSGeneral signs
Diffuse scleritis
Nodular scleritis
Necrotizing scleritis with inflammation
Necrotizing scleritis without inflammation
Posterior Scleritis
|
SYMPTOMSGeneral symptoms
Diffuse scleritis
Nodular scleritis
Necrotizing Scleritis with Inflammation
Necrotizing scleritis without Inflammation
Posterior scleritis
|
TREATMENT/MANAGEMENTNon-necrotizing scleritis
Necrotizing Scleritis or Non-necrotizing scleritis refractory to NSAIDs
Severe Necrotizing Scleritis and co-existing autoimmune disease
|
Quist TS, Vogelgesang S, Goins KM. Scleritis: A Case Report and Overview. EyeRounds.org. November 16, 2018. Available from https://eyerounds.org/cases/281-scleritis.htm

Ophthalmic Atlas Images by EyeRounds.org, The University of Iowa are licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License.

Address
University of IowaLegal
Related Links
