
Chief Complaint: Suspected chronic angle closure of the left eye (OS)
History of Present Illness: A 12-year-old female was referred to the Glaucoma service for suspected chronic angle closure OS. Intraocular pressures were measured to be 19 mm Hg in the right eye (OD) and 38 mm Hg OS with concern for a very shallow anterior chamber OS by an outside optometrist. She was started on brimonidine-timolol 0.2-0.5% twice daily (BID) OS prior to evaluation here.
At presentation, she reported difficulty focusing on the board at school, needing to hold things very close to her face to focus, and difficulty noticing people standing beside her.
Past Ocular History:
Past Medical History:
Medications:
Allergies: No known allergies
Family History: No family history of congenital cataracts, glaucoma, or other eye diseases
Social History: Non-contributory
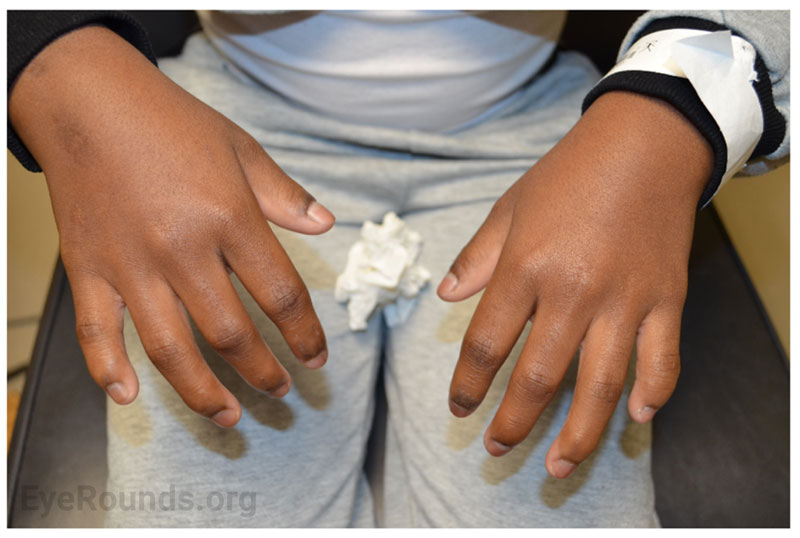
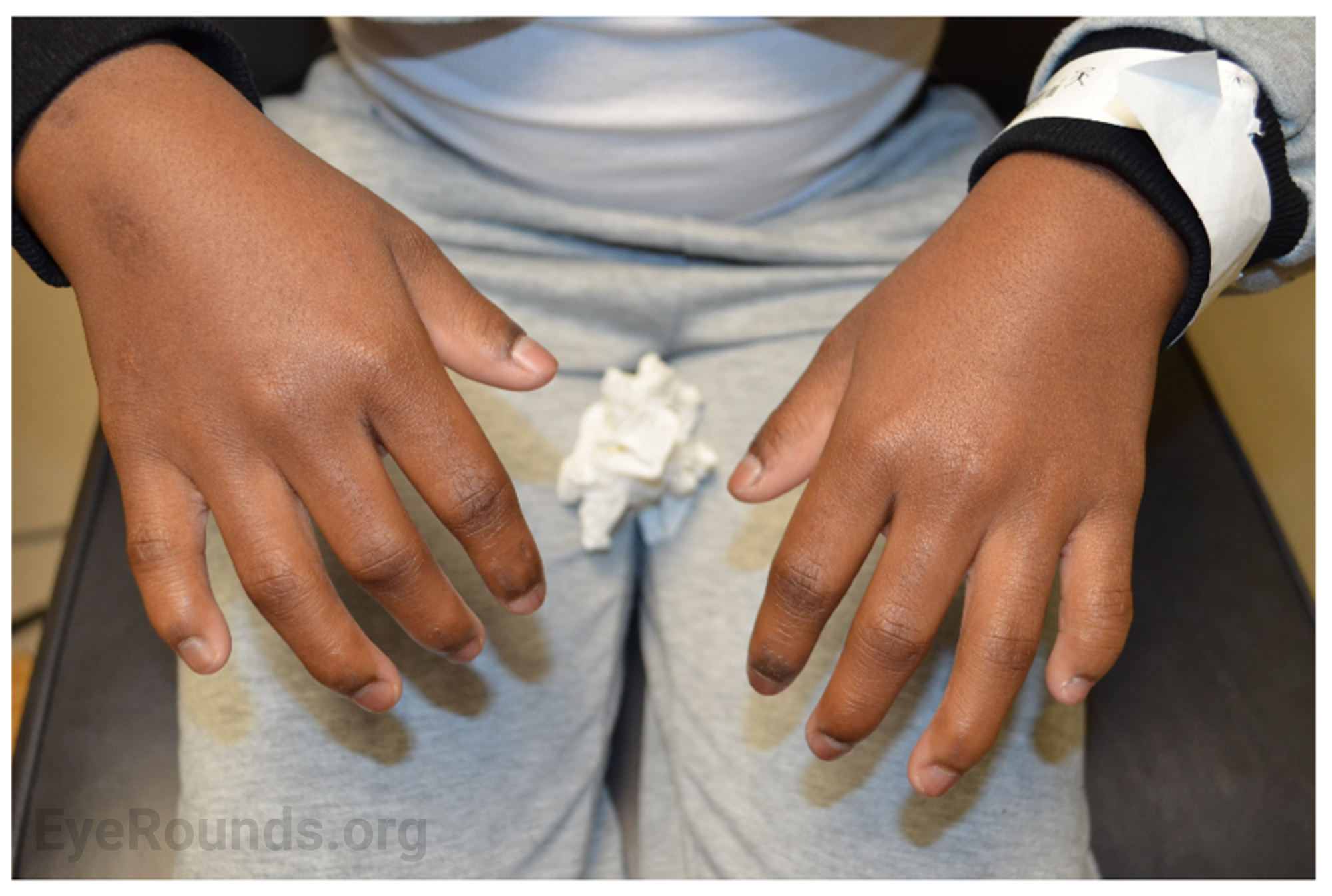
Review of Systems: Positive for finger stiffness and difficulty making a fist bilaterally. All other systems reviewed and negative.
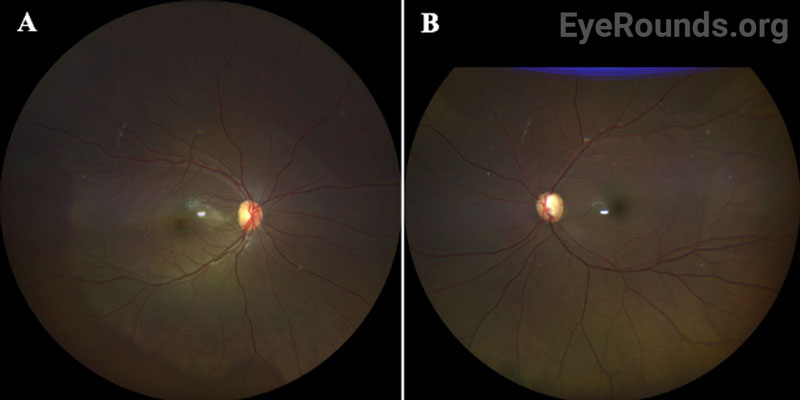
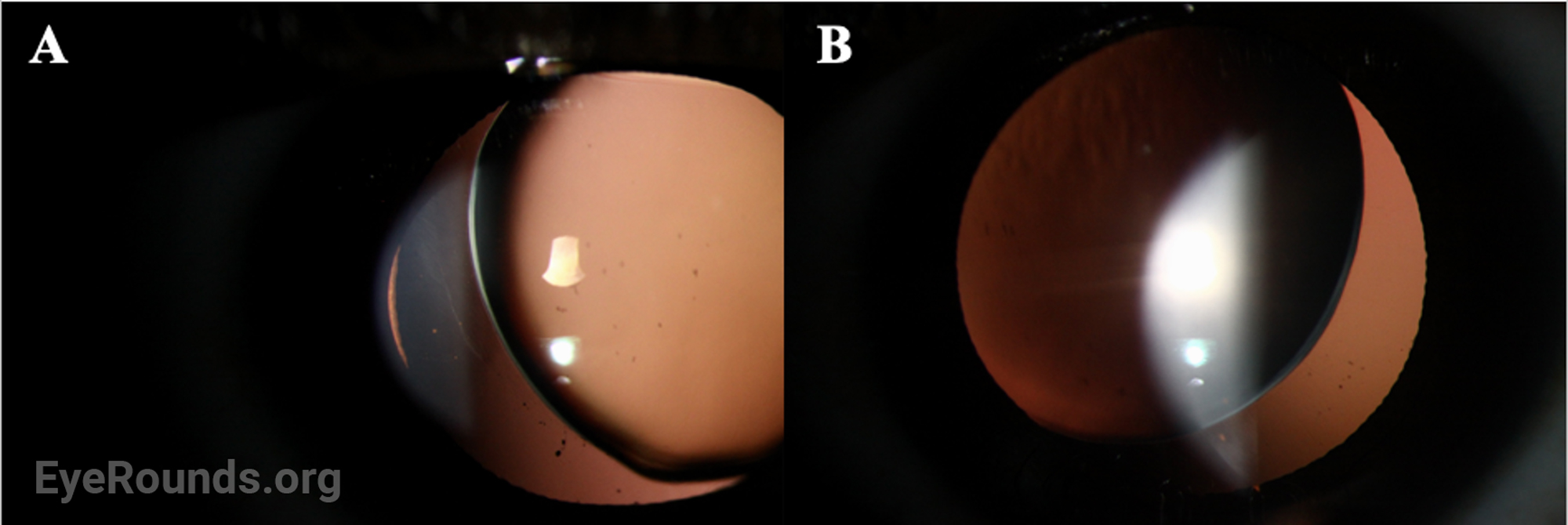
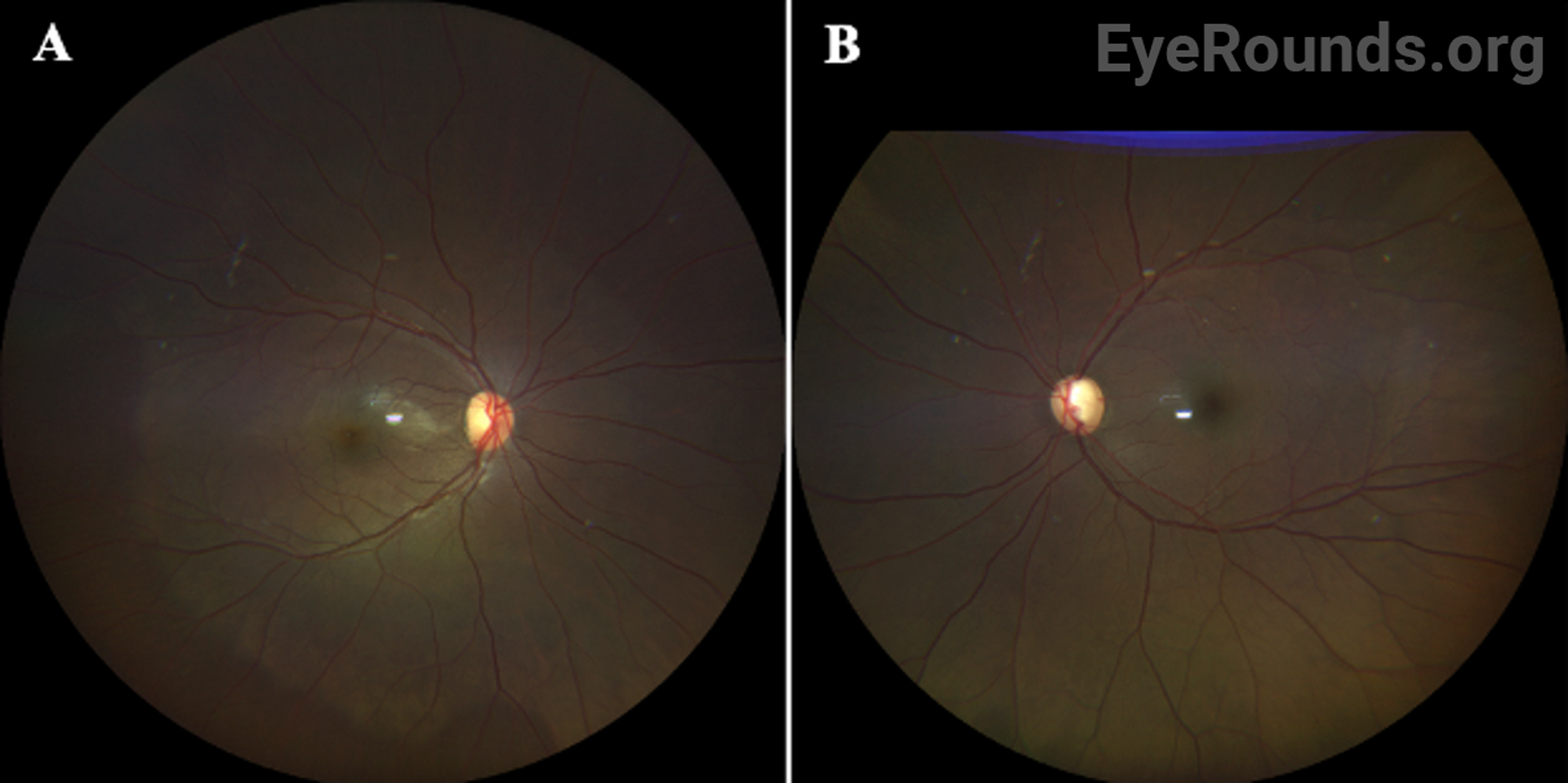
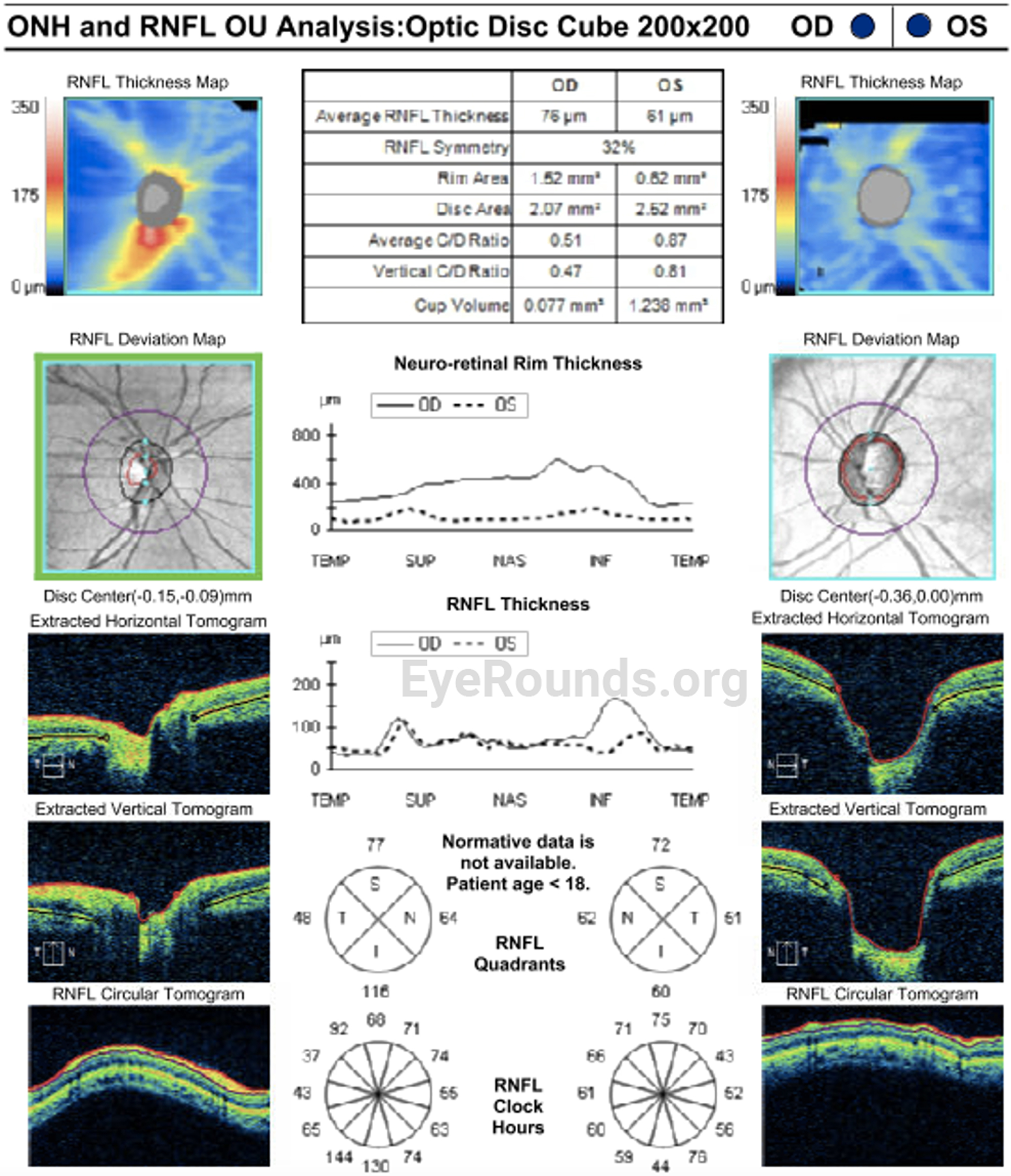
OCULAR EXAMINATION
Differential Diagnosis for a Dislocated Lens
CLINICAL COURSE
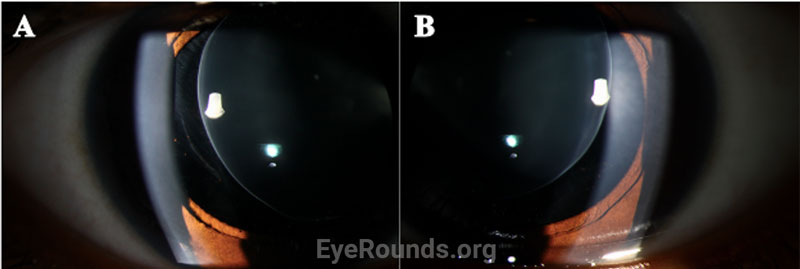
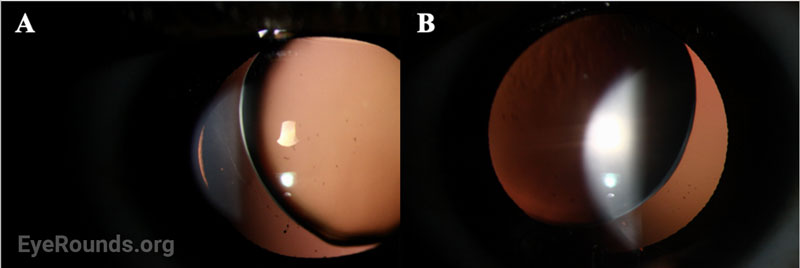
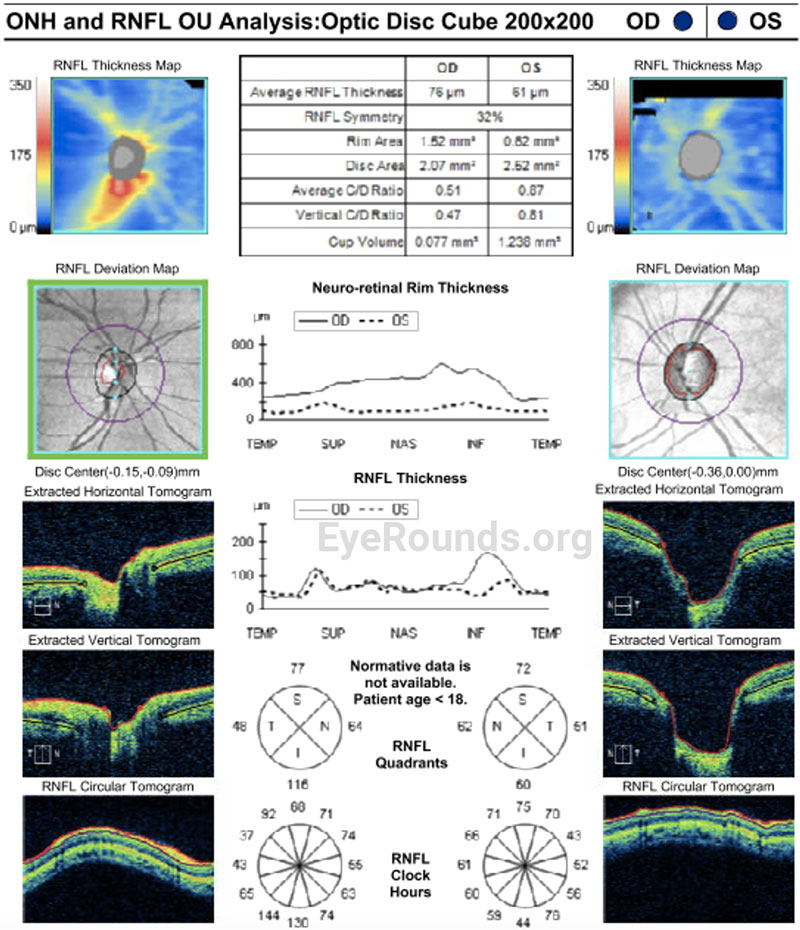
Given the clinical findings of bilateral spherophakia with lens dislocation, high myopia without axial elongation, narrow angle glaucoma based on RNFL thinning and disc changes as listed above, short stature, brachydactyly, and joint stiffness as well as the patient’s history of congenital heart disease, the patient was diagnosed with Weill-Marchesani Syndrome. The patient successfully underwent lensectomy with anterior vitrectomy, left eye first followed by the right eye. The patient was left aphakic, as the main concerns at the time were zonular instability and glaucoma progression. The patient and her family will consider a secondary intraocular lens in the future. Intraocular pressures remained normal. The patient was referred for a genetics evaluation given her syndromic features.
DIAGNOSIS: Weill-Marchesani Syndrome
DISCUSSION
Overview
Weill-Marchesani Syndrome (WMS), also known as spherophakia-brachymorphia syndrome or congenital mesodermal dysmorphodystrophy, is a rare connective tissue disorder that typically presents during childhood and is characterized by lens abnormalities (microspherophakia, ectopia lentis, and lenticular myopia), secondary glaucoma, short stature, brachydactyly, joint stiffness, and cardiovascular defects [2-5].
Epidemiology
The prevalence of WMS is estimated to be 1:100,000 in the general population [2,3].
Genetics
WMS can occur via autosomal recessive (AR) inheritance (45%), autosomal dominant (AD) inheritance (39%), or sporadically (16%) [2,3,5,6]. Penetrance is thought to be 100% for both AD and AR forms. Intra- and inter-familial expressivity varies [3]. The clinical manifestations and disease severity of WMS do not differ between AR, AD, or sporadic forms [6].
Four genes encoding extracellular matrix components are implicated in the pathogenesis of WMS. Pathogenic variants of the FBN1 gene are inherited in an AD manner (WMS 2). Pathogenic variants of the ADAMTS10, ADAMTS17, and LTBP2 genes are inherited in an AR manner (WMS 1, WMS 4, and WMS 3, respectively) [2,3].
Pathophysiology
Fibrillin, a glycoprotein encoded by the FBN1 gene, is a component of many connective tissues throughout the body, including blood vessels, ligaments, dermis, and ciliary zonules. In the ciliary zonules, fibrillin serves as a structural scaffold of extensible microfibrils [2,3,5]. The ADAMTS (A Disintegrin and Metalloprotease with Thrombospondin Motifs) gene superfamily codes for several zinc metallopeptidases involved in proteoglycan processing and appears to play a role in fibrillin regulation and biogenesis [2,3]. Latent transforming growth factor beta-binding protein 2, encoded by the LTBP2 gene, is an extracellular matrix protein involved in the formation and longevity of ciliary zonule fibrillin-containing microfibrils [2,3]. Inherited mutations in the aforementioned genes that encode for microfibrils and zinc metallopeptidases, therefore, confer abnormalities in the structure and function of the lens-zonule apparatus.
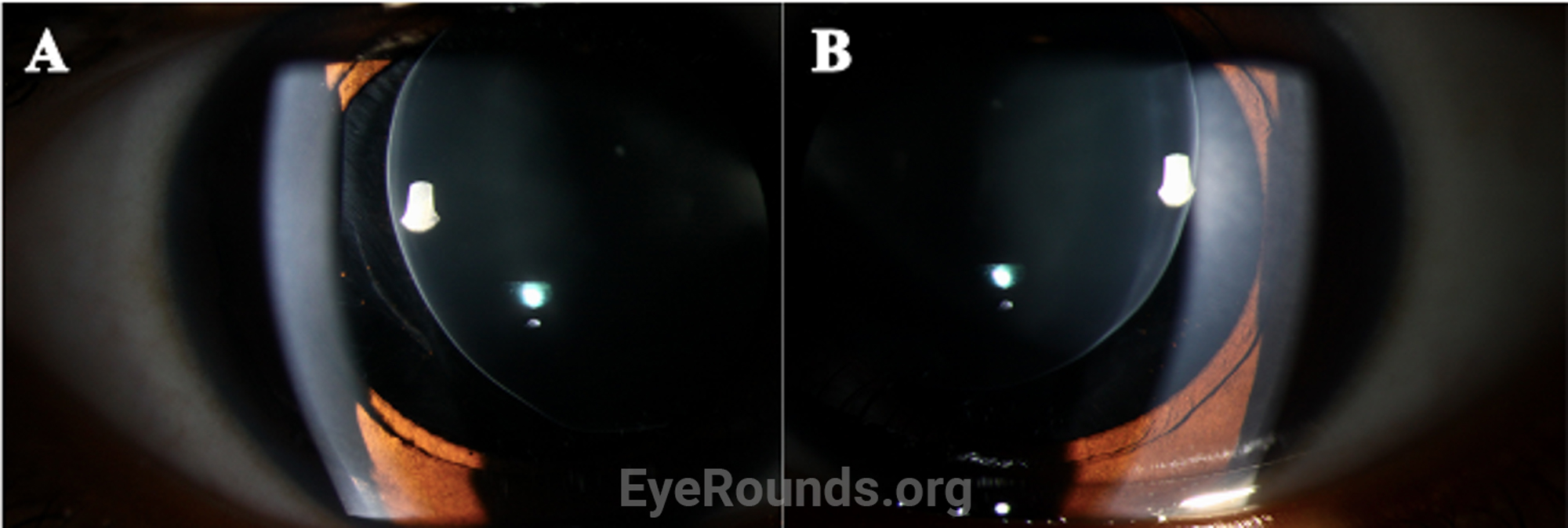
Disruption of fibrillin or fibrillin-associated proteins results in zonular weakness leading to lens hypermobility and spherophakia [2,3,5,7]. Hypermobile lenses are prone to dislocation and pupillary block [2,3,5,7,8]. Lens dislocation results in an acute decrease in visual acuity due to refractive shift. Pupillary block causes elevations in intraocular pressure that can lead to acute or chronic glaucomatous damage [2,3,7,8]. High myopia occurs as a result of the increased refractive power of the hyper-spherical lens. Of note, it is very unusual for an individual with high myopia to develop angle closure, as myopic eyes are often elongated with a deep anterior chamber [7,9]. In contrast, myopic individuals with WMS typically have a normal AEL and shallow to normal anterior chamber depth due to the mobile, spherical lens shifting the iris anteriorly.
Signs/Symptoms
Ocular manifestations of WMS include microspherophakia with consequent moderate to severe lenticular myopia, lens hypermobility with susceptibility to lens dislocation and pupillary block, and secondary glaucoma. In a review of 111 microspherophakic eyes, Senthil et al. reported a mean refractive error of -13.31±5.12 D [8]. Additionally, Razeghinejad et al. identified increased central corneal thickness as a feature of WMS [10]. Lenticular myopia is often the first ophthalmologic problem recognized with a mean age of presentation of 7.5 years [2,3]. In a review of 128 patients with WMS, Faivre et al. reported an 80% frequency of glaucoma [6].
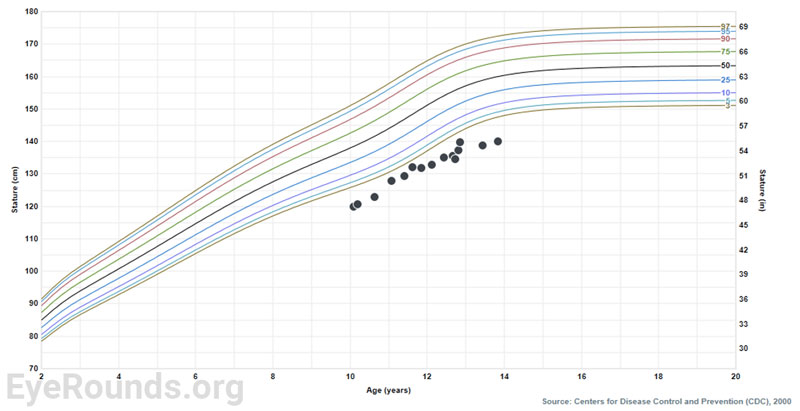
Musculoskeletal manifestations of WMS include short stature, brachydactyly, progressive joint stiffness, prominent interphalangeal joints, and pseudomuscular build [2,3]. Adult males and females with WMS are expected to achieve a height of 142-169 cm (4.7-5.5 ft.) and 130-157 cm (4.3-5.2 ft.), respectively [3]. Patients with WMS may also present with maxillary hypoplasia. Radiographic findings include short tubular bones, delayed bone age, and broad proximal phalanges [2,3].
Cardiovascular of WMS include patent ductus arteriosus, pulmonary stenosis, aortic stenosis, mitral valve prolapse, thoracic aortic aneurysm, cervical artery dissection, and prolonged QT interval [2,3].
Other manifestations include taut skin with thickened skin folds and mild intellectual disability [2-4].
A notable comparison is that between WMS and Marfan syndrome. Both diseases are the result of disruptions of fibrillin and fibrillin-associated proteins. As such, one might suspect that the manifestations of these disease would be similar. However, being an acromelic dysplasia, WMS has been described as “the opposite of Marfan syndrome” [11,12]. Though both diseases present with cardiovascular abnormalities, high myopia, and ectopia lentis, individuals with Marfan syndrome are typically tall, hypomuscular, hypermobile, and have arachnodactyly [5,12]. The prevalence of Marfan syndrome is estimated to be 1:3,000-5,000, making it far more common than WMS [12]. In a review of 396 cases of congenital ectopia lentis, Fuchs and Rosenberg found that 68.2% were due to Marfan syndrome, while only 0.73% were due to WMS [13]. Additionally, the myopia associated with Marfan syndrome is attributed to axial elongation of the eye rather than increased refractive power of the lens [5].
Diagnosis
There are no consensus clinical diagnostic criteria for WMS to date. The diagnosis should be suspected in individuals with characteristic clinical and radiographic findings and can be established with genetic evaluation.
For individuals with suspected AD WMS, genetic evaluation may include serial single-gene analysis of FBN1 to identify intragenic variants. If the inheritance of the disease is unclear, a multigene panel including FBN1, ADAMTS10, ADAMTS17, and LTBP2 may be ordered to identify the responsible genetic variant. If the presenting phenotype is not readily distinguishable from other inherited connective tissue disorders, comprehensive genomic testing (most commonly exome sequencing) may help clarify the diagnosis [2,3].
Management
Management of WMS focuses on treatment of ophthalmic and systemic manifestations of the disease.
Annual ophthalmic examination is recommended for early detection of ectopic lenses, pupillary block, and secondary glaucoma. Miotic and mydriatic agents should be avoided, as they may induce pupillary block [2,3]. Early lensectomy of the microspherophakic lens may improve visual acuity, prevent pupillary block, improve intraocular pressures, and prevent the development of secondary glaucoma. Management of more severe glaucoma may involve peripheral iridectomy or filtering surgery.
Management of systemic symptoms may include physical therapy to maintain joint mobility and regular cardiac examination to screen for anomalies. Patients should be counseled on the risk of participating in contact sports. Genetic counseling may be offered to patients and at-risk family members.
EPIDEMIOLOGY
ETIOLOGY
|
SIGNS
|
SYMPTOMS
|
TREATMENT/MANAGEMENT
|
Najdawi W, Rodriguez S, Mortensen ZQ, Pouw AE, Larson SA. Weill-Marchesani Syndrome. EyeRounds.org. Posted July 10, 2023; Available from https://EyeRounds.org/cases/344-weill-marchesani-syndrome.htm

Ophthalmic Atlas Images by EyeRounds.org, The University of Iowa are licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License.

Address
University of IowaLegal
Related Links


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}