
Chief complaint: left orbital mass
History of Present Illness: The patient is a 53-year-old male with a history of bilateral enucleation for retinoblastoma who presented to oculoplastics clinic because of a bump along his left upper eyelid. The mass had been present for the past month and was accompanied by mild discharge, but no pain. The patient’s ocular history is significant for bilateral retinoblastoma diagnosed at the age of 2 years, for which he underwent bilateral enucleation followed by 6 weeks of radiation. The patient is high functioning and navigates with a Seeing Eye dog.
Past Ocular History: See History of Present Illness
Medical History: Bilateral retinoblastoma, hypothyroidism, hyperlipidemia
Medications: Lovastatin, levothyroxine
Allergies: None
Family History: Unremarkable for previous retinoblastoma
Social History: He is employed and has adapted well to his disability. He is married and has no children. The patient is self-employed, working with vending machines.
Review of systems: History of lipomas and sebaceous cysts removed from posterior scalp and posterior neck.
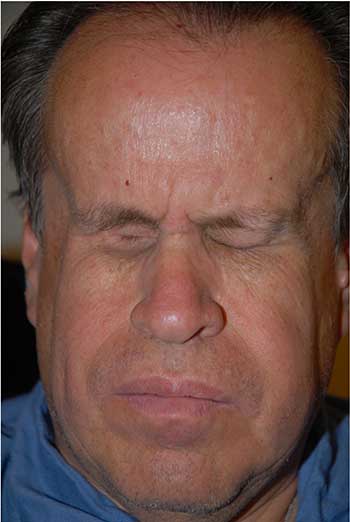
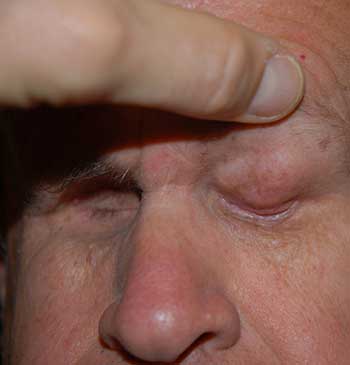
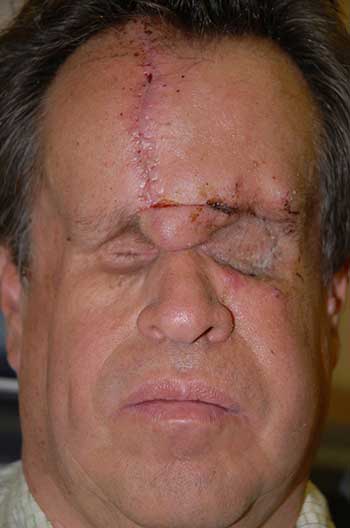
External: hourglass configuration of the head secondary to previous radiation therapy with severely contracted sockets (unable to wear prostheses) (Figure 1).
 |
 |
 |
 |
Diagnosis: Leiomyosarcoma of the left orbit secondary to radiation and germline retinoblastoma mutation 50 years after bilateral enucleation.
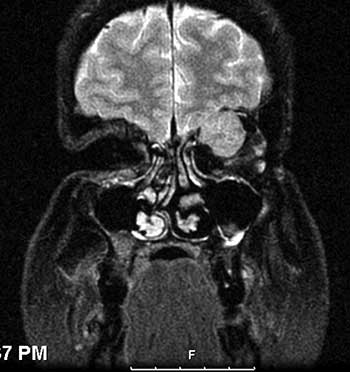
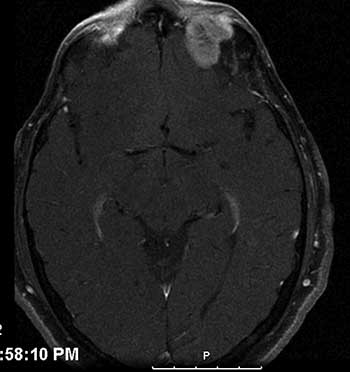
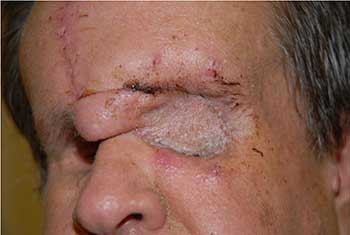
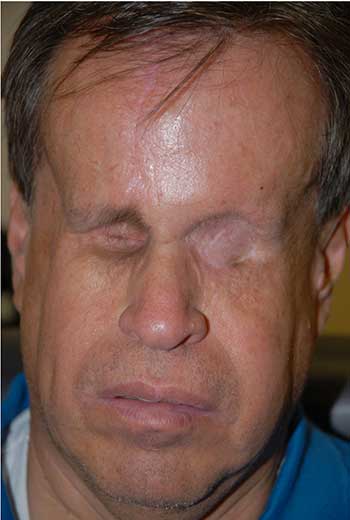
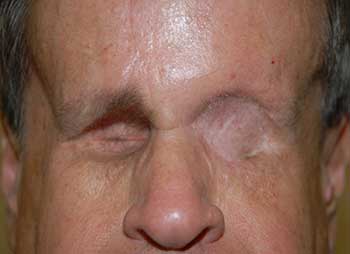
Course: The patient was diagnosed with a high-grade leiomyosarcoma of the left orbit, which was either encroaching or contiguous with the brain. PET/CT imaging showed no distant metastases. The patient underwent exenteration of the left orbit with preservation of the lower eyelid skin and orbicularis. The lesion margins were clear of tumor. In conjunction with this procedure, he also underwent neurosurgical resection of the dura with dural repair through the orbital roof. The skin was reconstructed with a median forehead flap and mobilization of the cheek (Figure 3). The forehead flap was then transected after six weeks. Following the surgery, the patient underwent adjuvant radiation therapy (image-guided radiation therapy of 60 Gy in 30 fractions). On the most recent follow-up one year after exenteration, there had been no recurrence (Figure 4).
 |
 |
 |
 |
Retinoblastoma is the most common primary intraocular malignancy of childhood, with an incidence of 1 in 15,000 live births (For a more detailed description and primary treatment for retinoblastoma, see Coombs and Boldt, Eyerounds 2005 [Coombs and Boldt, 2005]). Retinoblastoma is caused by a mutation in the retinoblastoma gene (RB1) located on the long arm of chromosome 13 at locus 14 (13q14), which was the first tumor suppressor gene cloned. RB1 encodes the p105 retinoblastoma protein, which is involved in regulating the cell cycle. Both copies of RB1 must be mutated for retinoblastoma to develop. While only 6% of retinoblastoma patients have a family history of retinoblastoma, bilateral retinoblastoma is almost always due to a germline mutation in RB1 often from a de novo mutation. In addition, 15% of patients with unilateral retinoblastoma are carriers of a germline RB1 mutation. In accordance with the Knudson two-hit hypothesis, patients with a germline mutation in retinoblastoma only require a single somatic mutation to develop retinoblastoma, which explains the tendency for patients with hereditary retinoblastoma to present early with bilateral multifocal lesions. Sporadic retinoblastoma requires two somatic mutations within the same retinal cell for tumor formation. Detecting carriers of germline mutations in retinoblastoma is not only important for genetic counseling, but also because germline mutations place patients at increased risk of secondary malignancies in the future, most commonly osteosarcoma, soft tissue sarcoma (especially leiomyosarcoma), melanoma, carcinoma, and brain cancer. Multiple longitudinal studies have demonstrated that survivors of hereditary retinoblastoma have an increased risk of developing secondary malignancies, while survivors of non-hereditary retinoblastoma are not at an increased risk of secondary tumors [Kleinerman, et al., 2005; Marees, et al., 2008; Roarty, et al., 1988].
The risk of developing a secondary malignancy in survivors of hereditary retinoblastoma is roughly 1% per year, ranging from 30-48% at 50 years [Kleinerman, et al., 2005; MacCarthy, et al., 2009]. Prior treatment with external radiation increases the risk of a secondary tumor 3-5 times, with higher risk in patients given radiation prior to 12 months of age. Two-third of tumors arise within the field of radiation, and one-third arise outside. Although prior radiation is a clear risk factor for secondary malignancies, patients without prior radiation are still at increased risk, with a cumulative risk of 21% at 50 years [Kleinerman, et al., 2005]. Recognizing the increased risk of secondary tumors in patients with hereditary retinoblastoma is important because the mortality rate from secondary malignancies associated with hereditary retinoblastoma is 25.5% at 50 years [Yu, et al., 2009]. This is far higher than the mortality from primary retinoblastoma, which has a survival rate approaching 99% in the developed world [Lin and O'Brien, 2009].
The pathogenesis behind the increased risk of secondary tumors in hereditary retinoblastoma is due to the germline mutation in RB1 gene. When initially described in retinoblastoma, RB1 was found to regulate the cell cycle by arresting cells in G1 through the inhibition of E2F transcription factors, the loss of which led to the development of retinoblastoma [Sellers and Kaelin, 1997]. Since these seminal findings, RB1 has been also found to also play a role in cellular differentiation, regulation of apoptosis, and preservation of chromosomal stability [Burkhart and Sage, 2008]. In addition to retinoblastoma, RB1 mutations have been shown to play a role in multiple kinds of tumors, including sarcomas, small cell lung cancer, bladder cancer, and breast cancer [Burkhart and Sage, 2008]. Loss of RB1 function contributes to both cancer initiation and progression likely through the interaction with multiple other tumor suppressor genes and oncogenes. In patients with hereditary retinoblastoma, the germline loss of one copy of the RB1 gene leads to an increased risk of future malignancies that is often not appreciated until many years after the diagnosis of hereditary retinoblastoma.
Leiomyosarcoma is the most common secondary soft-tissue sarcoma seen in hereditary retinoblastoma and is only second to osteosarcoma among all secondary malignancies [Kleinerman, et al., 2007]. Seventy eight percent of leiomyosarcomas occur 30 or more years after the diagnosis of retinoblastoma [Kleinerman, et al., 2007]. Leiomyosarcoma is rarely seen in the orbit, but is a known complication in patients with hereditary retinoblastoma, especially with prior radiation therapy. Orbital leiomyosarcoma can also develop from metastatic lesions from distant sites and from secondary extension from the paranasal sinuses [Kaltreider, et al., 1987]. Rarely, patients can develop primary leiomyosarcoma of the orbit not associated with a history of retinoblastoma; this typically occurs in older females with a mean age of is 63.8 years old [Lin, et al., 2005]. Orbital leiomyosarcoma most commonly presents as painless proptosis, but it can also present with decreased vision or as a palpable mass. These tumors are thought to arise from vascular smooth muscle or the Muller muscle. On histopathologic examination, leiomyosarcomas are composed of spindle-shaped cells with cigar-shaped nuclei. Giant cells, nuclear pleomorphism, hyperchromatism, and mitotic figures are common. Immunohistochemistry demonstrates desmin and smooth muscle actin immunoreactivity, which is consistent with smooth muscle tumors. Treatment for orbital leiomyosarcoma is surgical wide local resection, often requiring early orbital exenteration. Radiation and chemotherapy may be beneficial, but there have only been a few cases reported in the literature [Lin, et al., 2005].
EPIDEMIOLOGY
|
SIGNS
|
SYMPTOMS
| TREATMENT
|
Coombs J and Boldt HC. Retinoblastoma: 4 month old boy with a "blur" in one eye since birth according to his mother. EyeRounds.org. February 21, 2005; Available from: http://www.EyeRounds.org/cases/case27.htm. 2005.
Chen JJ, Allen RC. Secondary Tumors After Hereditary Retinoblastoma: A Case of Orbital Leiomyosarcoma 50 Years After Initial Enucleation and Radiation Therapy. EyeRounds.org. November 28, 2011; Available from: https://eyerounds.org/cases/142-leiomyosarcoma.htm

Ophthalmic Atlas Images by EyeRounds.org, The University of Iowa are licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License.

Address
University of IowaLegal
Related Links
