INITIAL PRESENTATION
Chief Complaint: Eye Shaking
History of Present Illness:
Patient is a 5-month-old male with a history of prematurity who was referred to the eye clinic for evaluation of nystagmus first noted by his parents and confirmed by his pediatrician. They have not noticed problems with him looking at faces, tracking objects, or any eye crossing or drifting. He has demonstrated normal growth and development since birth and has reached all developmental milestones appropriate for his age. No other visual concerns have been noted.
Past Ocular History:
Past Medical History:
Medications:
Allergies:
Family History:
Social History:
Review of Systems:
OCULAR EXAMINATION
| OD | OS | |
|---|---|---|
| Lids/lashes | Normal | Normal |
| Conjunctiva/sclera | Normal | Normal |
| Cornea | Clear | Clear |
| Anterior chamber | Deep and quiet | Deep and quiet |
| Iris | Normal | Normal |
| Lens | Clear | Clear |
| OD | OS | |
|---|---|---|
| Vitreous | Clear | Clear |
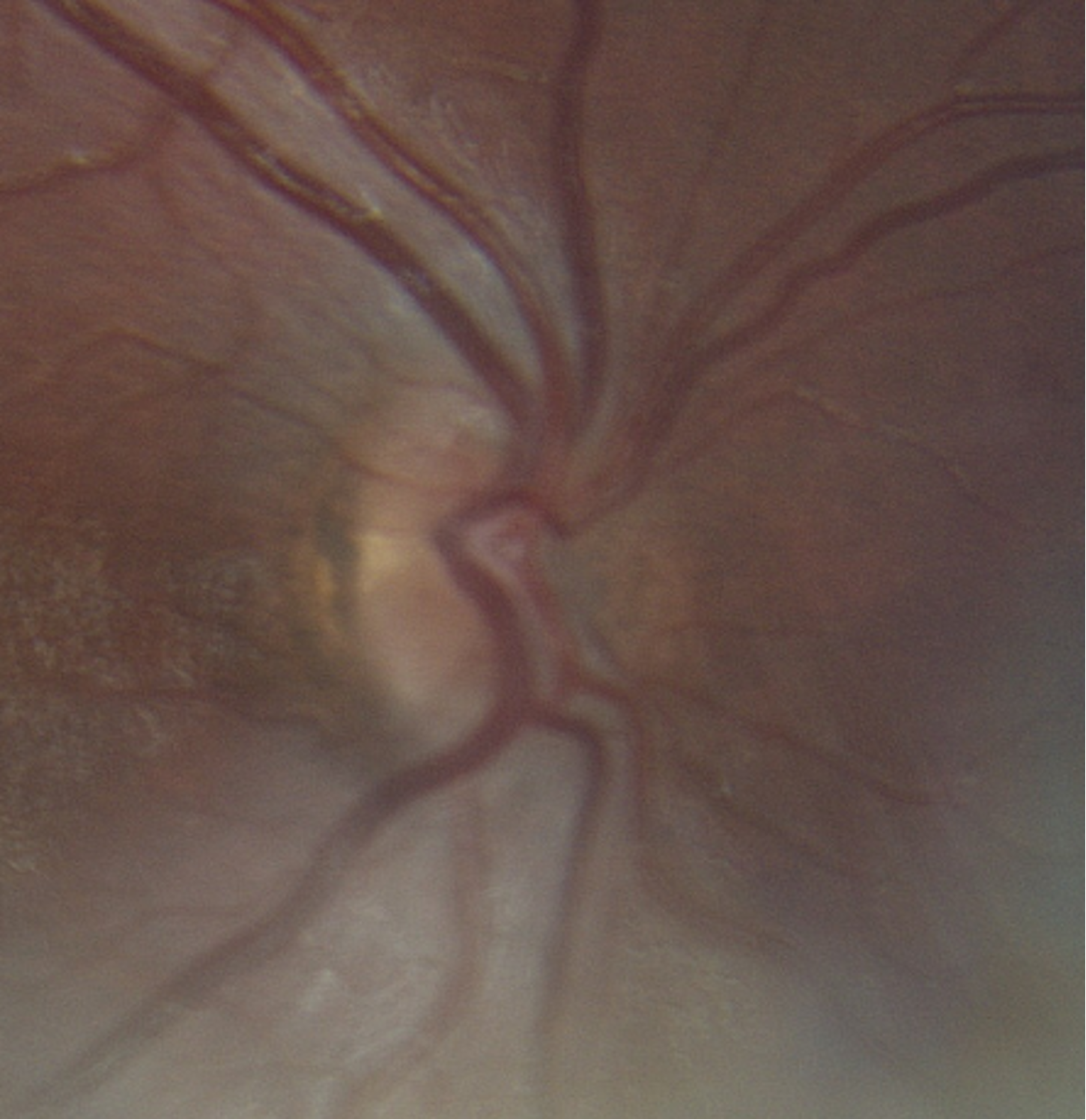
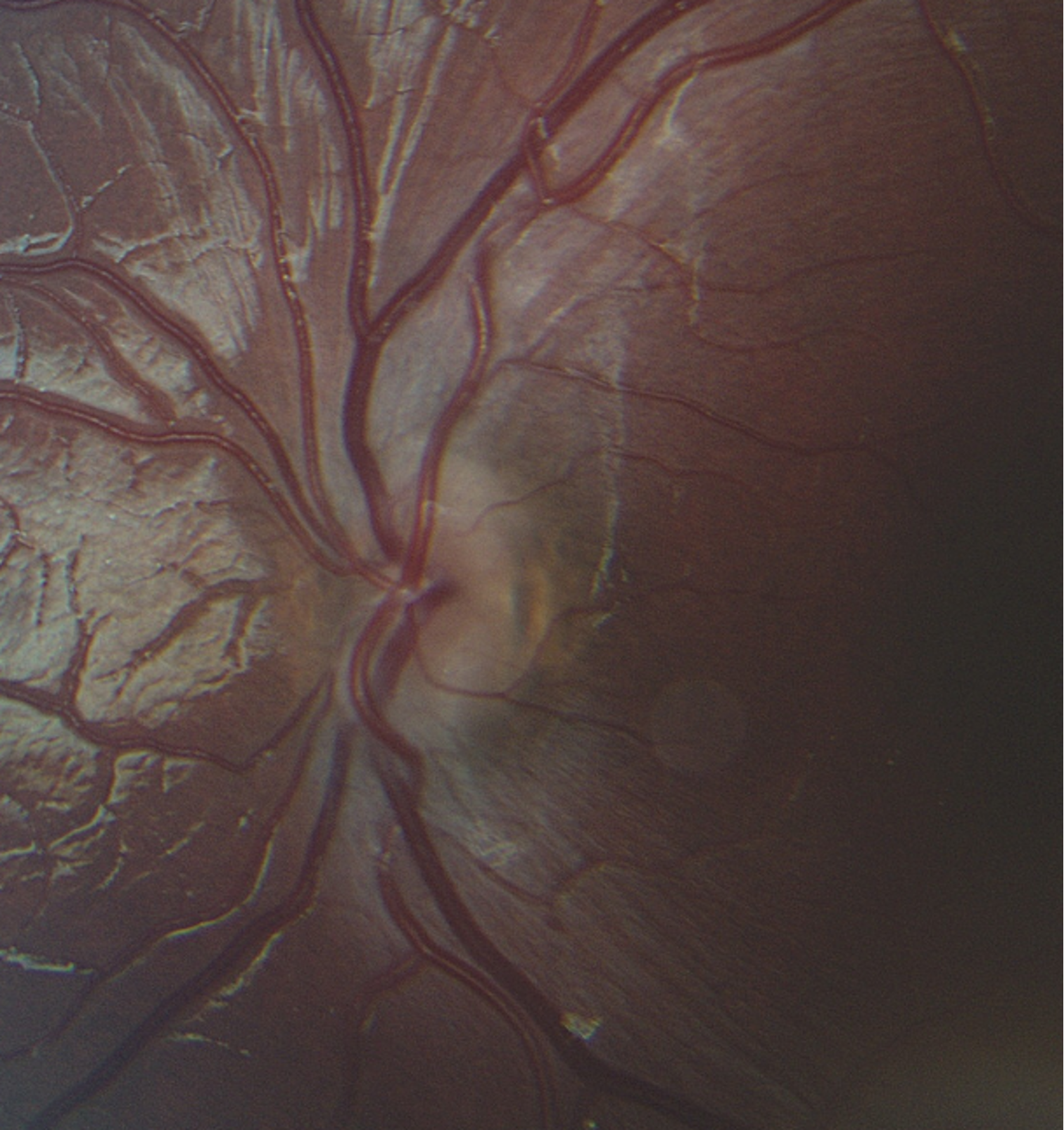
| Disc | Very small, normal color, no edema | Very small, normal color, no edema |
| Macula | Normal | Normal |
| Vessels | Normal | Normal |
| Periphery | Normal | Normal |
DIFFERENTIAL DIAGNOSIS:
CLINICAL COURSE
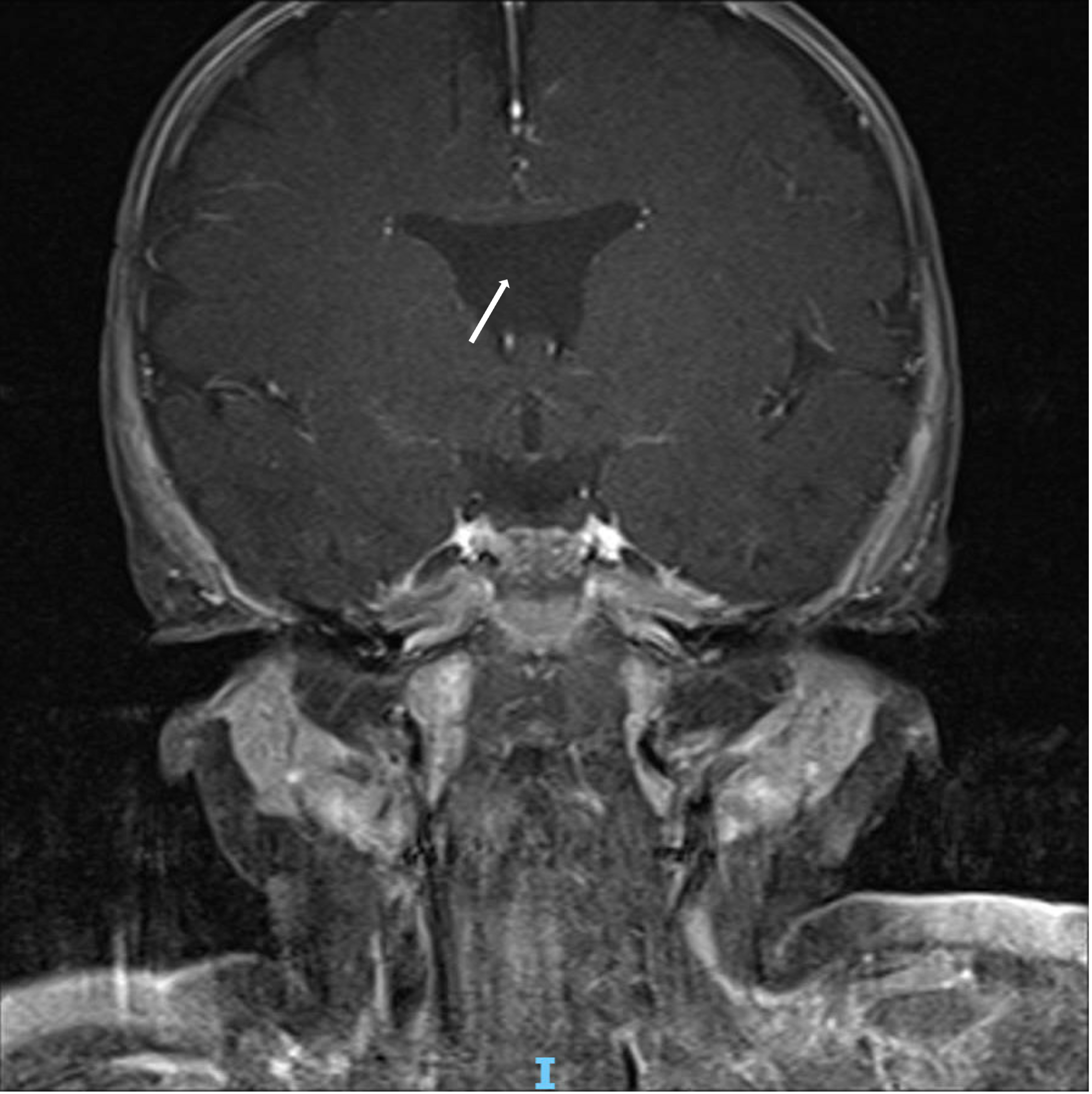
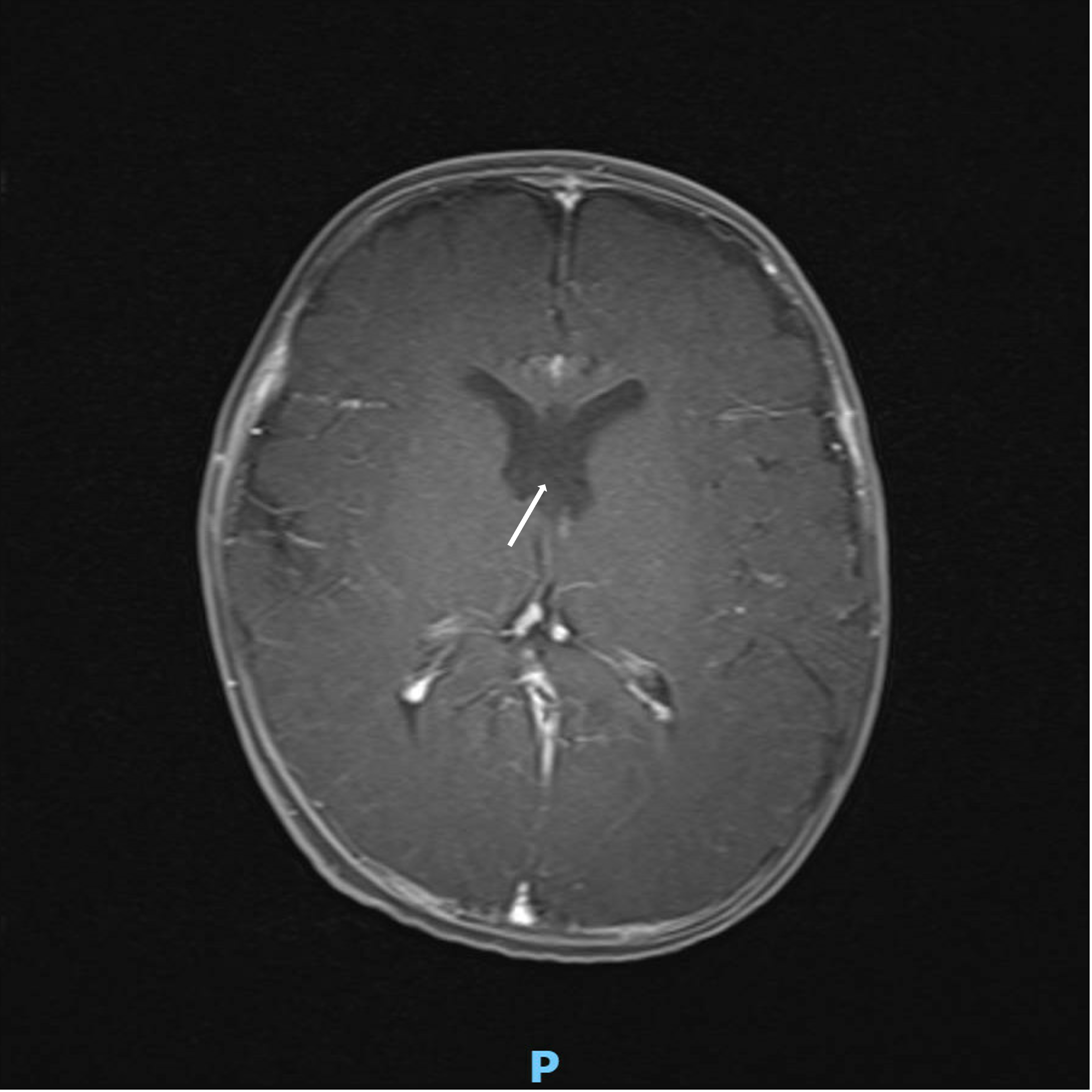
At the initial presentation, pituitary function labs and neuroimaging were ordered given the association of optic nerve hypoplasia with endocrine dysfunction and midline brain abnormalities. Neuroimaging demonstrated an absent septum pellucidum (Figure 1). The patient established with endocrinology and pituitary function was normal. Given the constellation of optic nerve hypoplasia and MRI brain result, he was diagnosed with septo-optic dysplasia.
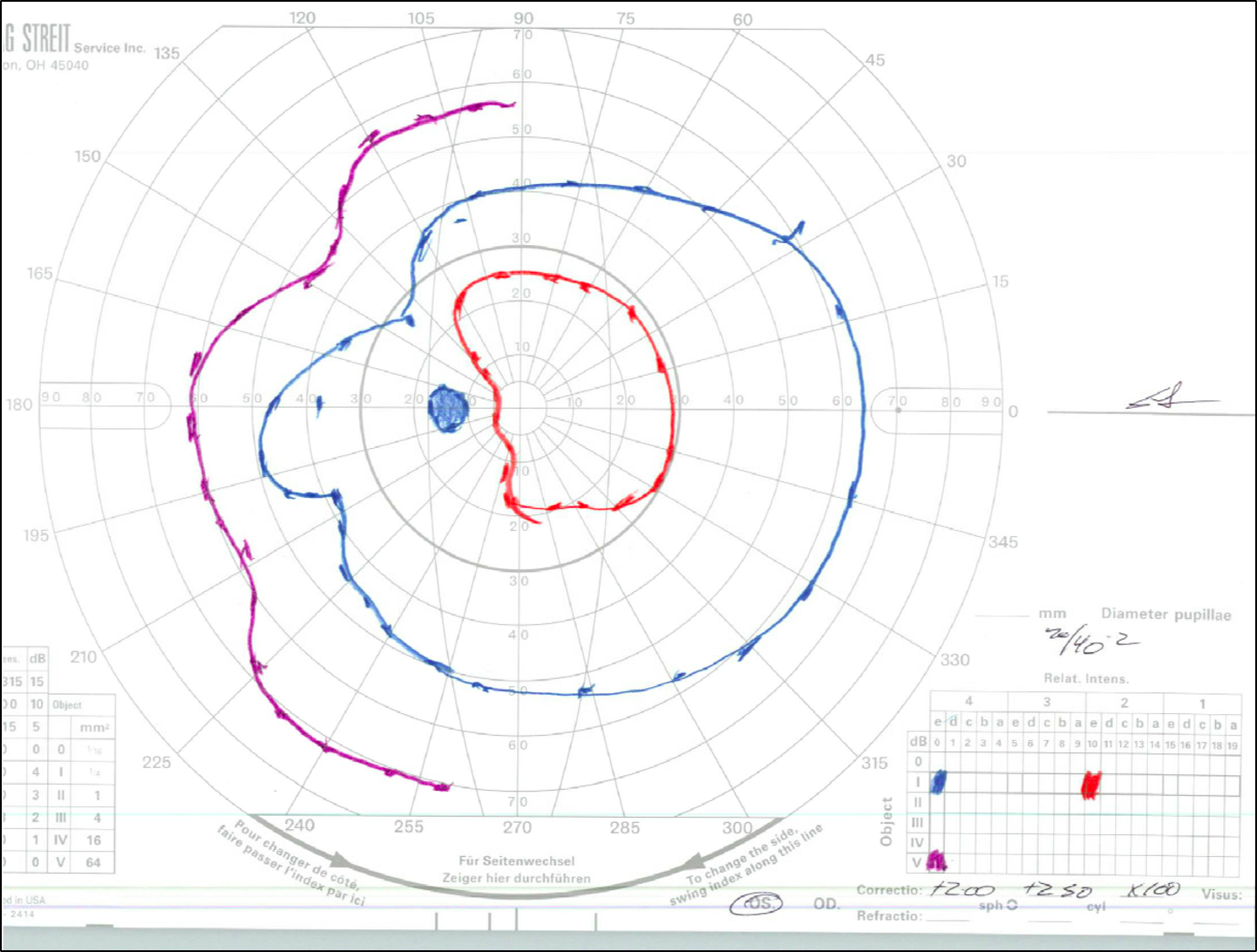
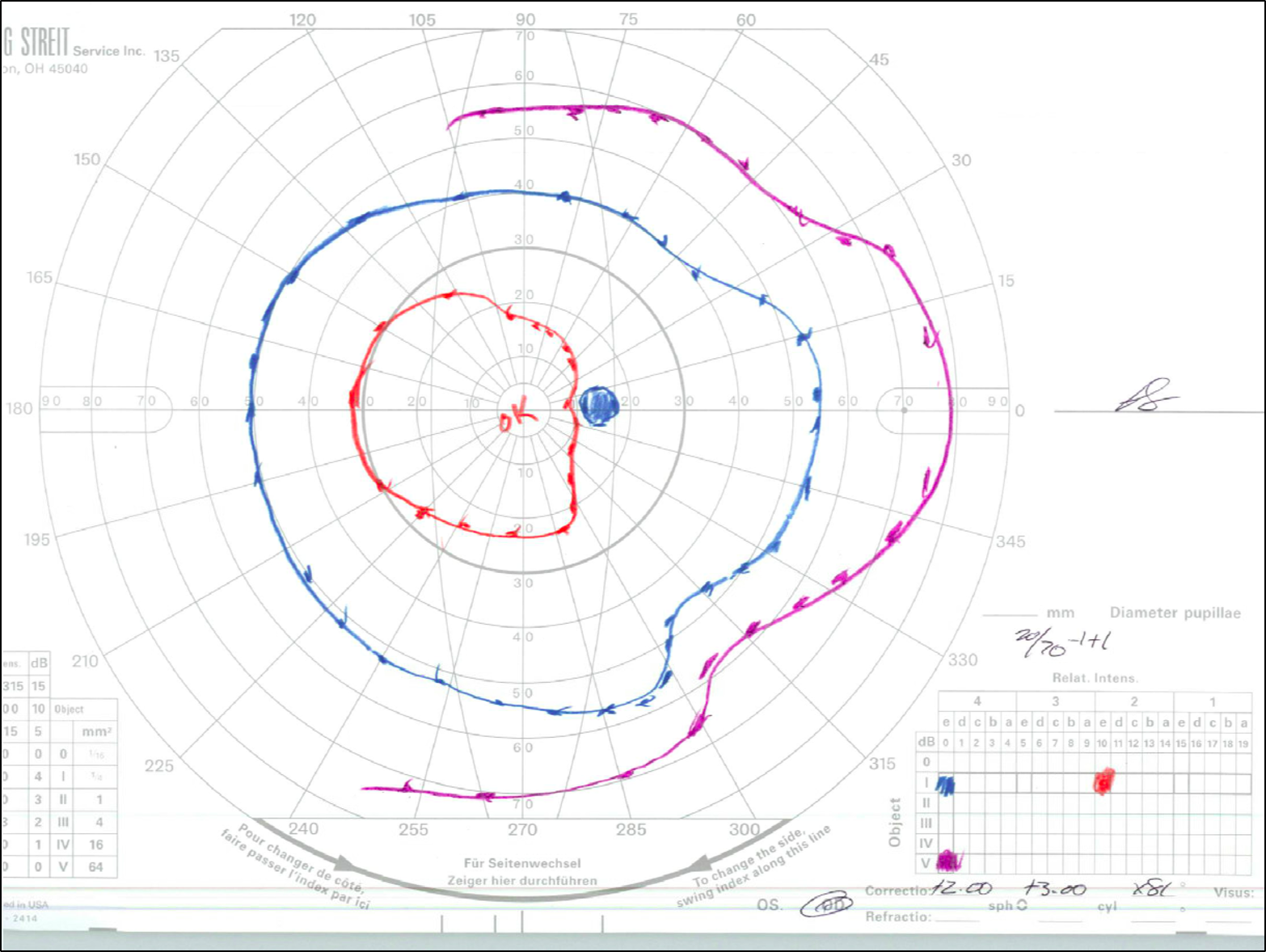
Goldmann Visual Field (GVF) testing was obtained when the patient was old enough to participate (Figure 3).
Over the course of multiple follow-ups, the patient has demonstrated stable vision. His most recent examination at the age of 16 years old showed:
DIAGNOSIS: Septo-Optic Dysplasia
DISCUSSION
Etiology
Septo-optic dysplasia (SOD) is a heterogeneous developmental disorder defined by the classic triad: optic nerve hypoplasia (ONH), midline brain abnormalities (absent septum pellucidum ± corpus callosum agenesis) and pituitary hypoplasia. It is a multifactorial condition with contributions from both genetic and environmental factors. The key genes implicated in this disease include HESX1, SOX2, SOX3, OTX2, SHH and ARID1A (1–4), which encode transcription factors critical in the development of the optic nerve, early forebrain, and pituitary gland. Thus, mutations in these genes can disrupt normal embryogenesis and lead to the classic features of the SOD triad, with ONH being the most consistent ocular finding.
However, even in confirmed genetic cases, phenotypic variability is high, suggesting environmental factors play a vital role in the development of this condition. Reported risk factors include young maternal age, primigravida status, and maternal diabetes. Teratogen exposure and viral infections during early gestation (5, 6), coinciding with critical periods of optic nerve, forebrain, and pituitary development, are also risk factors. These genetic and environmental factors contribute to the SOD presentation in patients.
Pathophysiology and Natural History:
The first and most consistent component of SOD is the hypoplastic optic nerves. ONH is characterized by a congenital reduction in the number of retinal ganglion cell axons, leading to a small optic disc and impaired transmission of visual information from the eyes to the brain. This results from disruption of early forebrain and optic nerve development during the first trimester of gestation. The natural history is nonprogressive, but visual impairment ranges from mild to severe and may be unilateral or bilateral.
The second and third components of the SOD triad are midline brain abnormalities and pituitary hypoplasia. Midline defects include agenesis or hypoplasia of the septum pellucidum and corpus callosum, arising from the disruption of early telencephalic development. Pituitary hypoplasia results from abnormal development of Rathke’s pouch and the ventral diencephalon, the embryonic precursors of the anterior and posterior pituitary (7, 8), respectively. Unlike ONH, which remains stable after birth, endocrine abnormalities may evolve over time, necessitating ongoing surveillance.
Signs/Symptoms/Findings:
The clinical manifestations of SOD are highly variable depending on which components of the triad are present. Ocular features from ONH are the most consistent and include decreased visual acuity, visual field defects, nystagmus, strabismus, and refractive error. The severity of visual acuity loss is determined by the degree of retinal ganglion cell axon loss present at birth. However, functional outcomes may improve modestly with refractive correction and amblyopia therapy. Children with bilateral ONH are at particularly high risk of legal blindness (9, 10). On physical examination, optic nerve hypoplasia is characterized by a small optic disc, often with the classic double-ring sign. The disc-macula to disc-diameter ratio is typically increased due to the underdeveloped optic nerve head (11).
In addition to the ocular features, patients may present with neurological findings in patients with midline brain abnormalities (10, 12–14), such as seizures, developmental delay, or motor deficits. Endocrine abnormalities have also been reported related to pituitary hypoplasia, including growth hormone deficiency, adrenal insufficiency, hypothyroidism, and hypogonadism. These deficits may lead to growth failure, delayed puberty, life-threatening hypoglycemia, and adrenal crises if left untreated (9, 15).
Diagnosis:
The diagnosis of SOD is established when two of the three classic features are present. In the case of our patient, two of the three features were identified, confirming the diagnosis. Diagnosis typically involves a combination of ophthalmic examination to identify optic nerve hypoplasia, neuroimaging (MRI) to evaluate for midline brain defects, and endocrine testing to assess pituitary hormone function (15).
Treatment/Management:
Because manifestations may evolve over time, patients require multidisciplinary evaluation and ongoing follow-up with ophthalmology, neurology and endocrinology. From an ophthalmic standpoint, ONH requires regular monitoring of visual acuity to track developmental progress and detect amblyopia. Refractive error should be corrected early with glasses, and amblyopia therapy initiated as appropriate. Patients with strabismus may benefit from amblyopia therapy and/or surgical correction (16, 17). While visual impairment from ONH itself is nonprogressive, early intervention with these measures can optimize functional vision and prevent secondary vision loss.
With regard to midline brain abnormalities, management may involve antiepileptic medications for seizures, as well as physical therapy, occupational therapy, and early educational interventions to address motor and developmental delays. For patients with pituitary and endocrine dysfunction, hormone replacement may be necessary for deficiencies in growth hormone, thyroid hormone, cortisol, and sex hormones. Because endocrine deficits can emerge over time, regular surveillance is recommended even in patients with normal baseline testing.
EPIDEMIOLOGY
|
ETIOLOGY
|
SIGNS/SYMPTOMS/EVALUATION
|
DIAGNOSIS/TREATMENT
|
Somisetty A, Anibire O, De Andrade LM. Septo-optic dysplasia. EyeRounds.org. May 13, 2026. Available from https://EyeRounds.org/cases/378-septo-optic-dysplasia.htm

Ophthalmic Atlas Images by EyeRounds.org, The University of Iowa are licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License.

Address
University of IowaLegal
Related Links