

RPE65-associated Leber Congenital Amaurosis
February 16, 2010
Chief Complaint: Poor fixation at 4 months of age
History of Present Illness: This 7-year-old female was originally seen at 4 months of age when her parents became concerned that she would not follow faces well. She would only follow bright lights. At her initial exam, she was found to have seesaw nystagmus. She had pink optic nerves with some retinal vessel attenuation. An electroretinogram (ERG) was performed which showed no detectable signal consistent with Leber congenital amaurosis (LCA).
She continued to be followed for several years during which time her nystagmus and visual perceptual ability improved. At her last visit, she was stable and doing well in school.
Past Ocular History: Myopia. No history of strabismus.
Medical History: Febrile seizures as an infant.
Family History: No history of blindness in the family. The patient s parents and two sisters have normal vision.
Physical Exam:
- Visual Acuity (with correction):
- Right Eye (OD): 20/200
- Left Eye (OS): 20/200
- Spectacles:
- OD: -8.50 +1.25 x 088
- OS: -9.00 +1.75 x 090
- Extraocular motility: Mild seesaw nystagmus
- Pupils:
- 6 mm to 4 mm in both eyes, sluggish reaction in both eyes.
- External and anterior segment examination: Normal
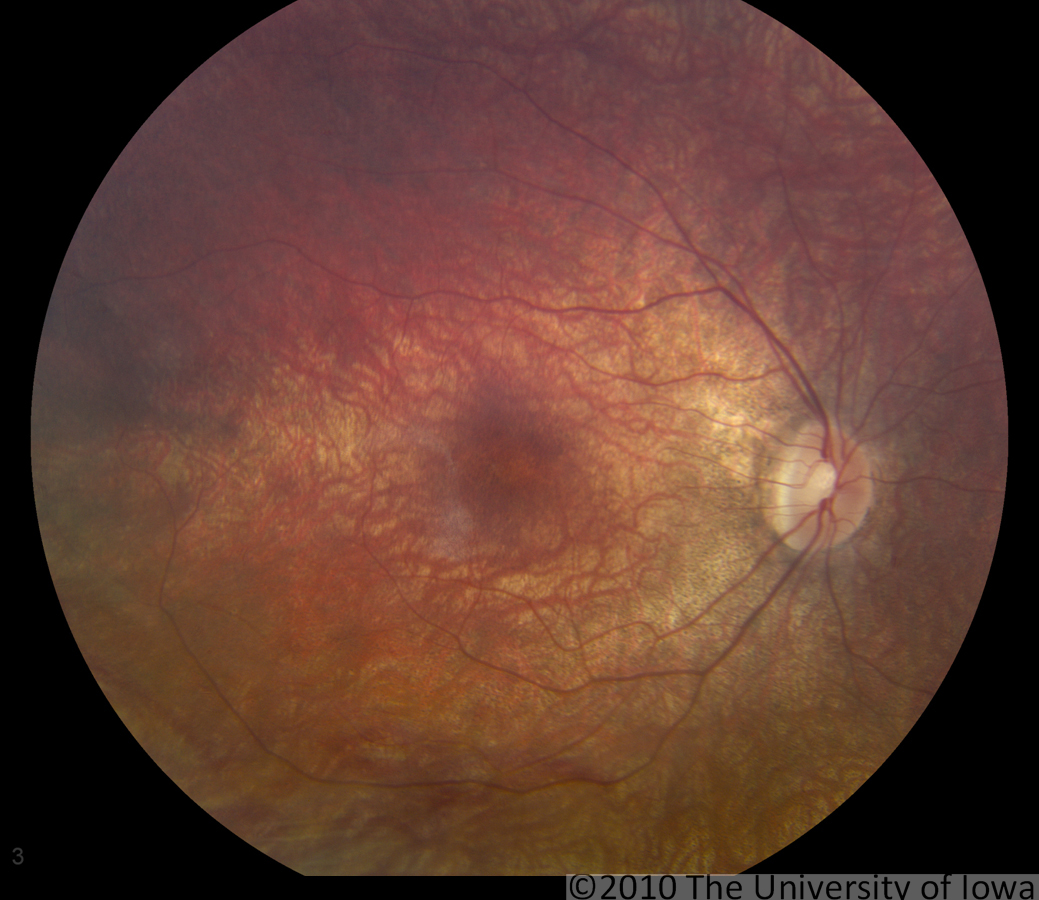
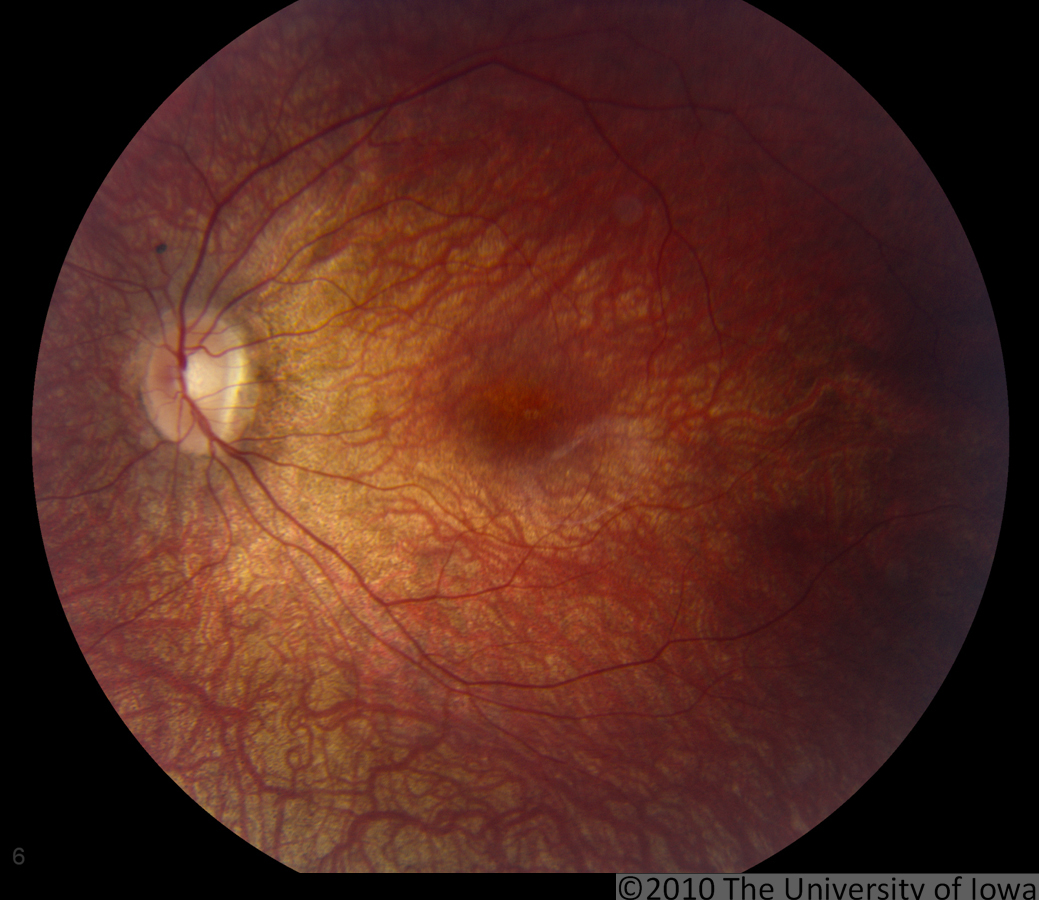
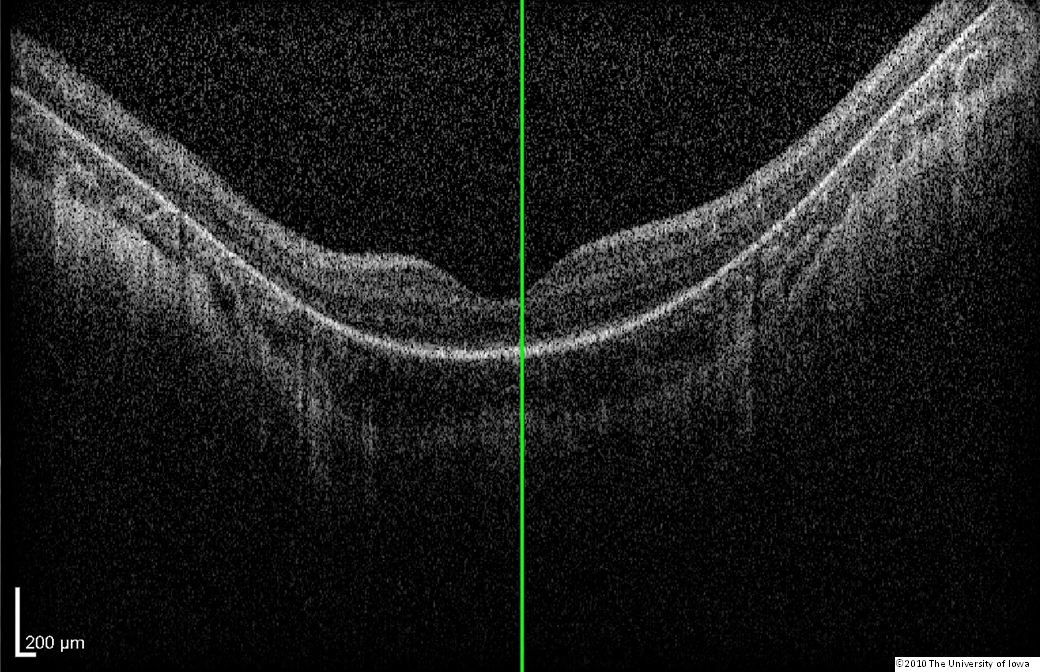
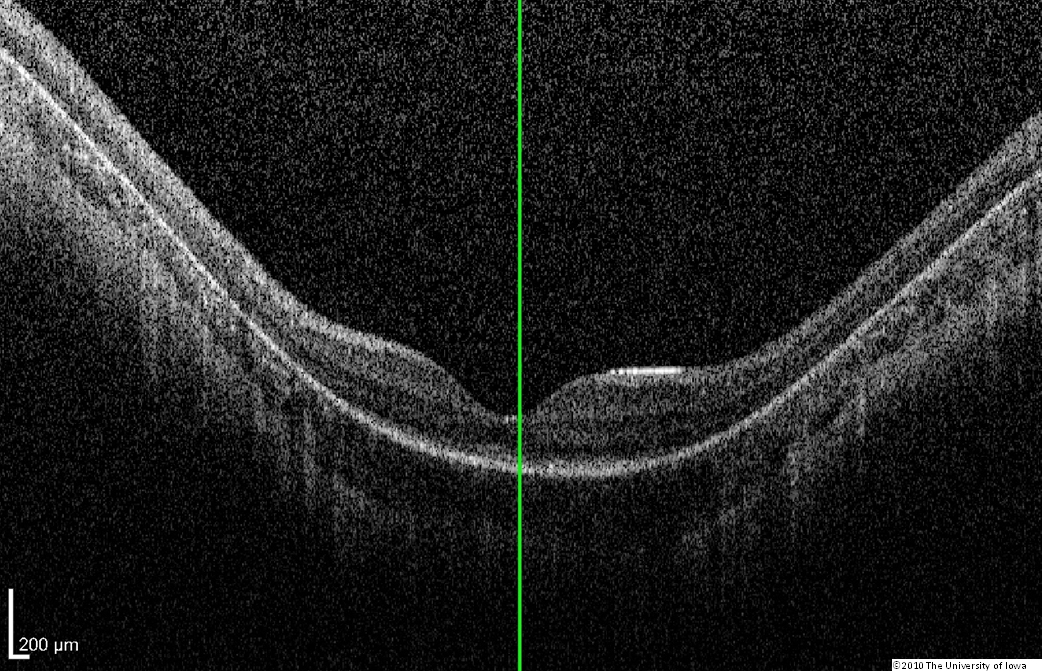
Dilated fundus exam: Both optic nerves appeared relatively pink with an intact neuroretinal rim and a cup/disc ratio of 0.3. There was a staphylomatous appearance to the macula in both eyes. Punctate yellow dots and associated pigment mottling were present in the macula and extended into the periphery of both eyes. The vessels were of normal caliber in both eyes. (See Figures 1-4)
 |
 |
| click on image for enlarged image | |
 |
 |
| click on image for enlarged image | |
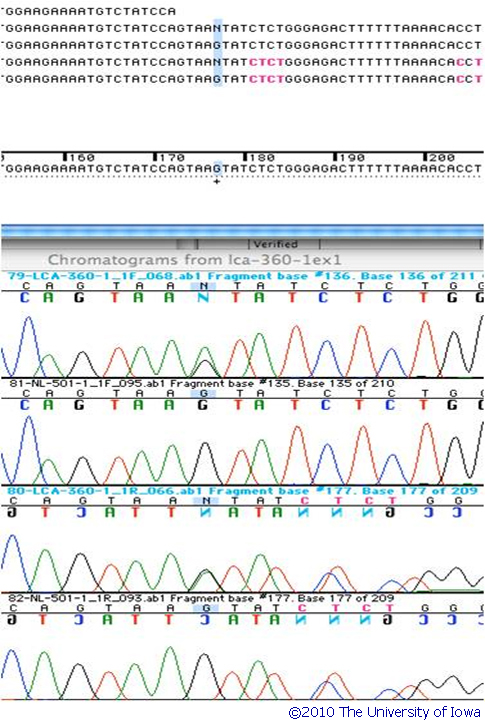
Genetic Testing: This patient had molecular genetic testing at the John and Marcia Carver Nonprofit Genetic Testing Laboratory (www.carverlab.org). Two disease-causing genetic variations were identified in her RPE65 gene. One is a heterozygous 1 base pair deletion of an A at codon 297 and the other is a heterozygous G>A nucleotide substitution at position +5 in intron 1. Both of these mutations have an estimate of pathogenic probability (EPP) of 3. The IVS1+5 variation is one of the most common disease-causing alleles in the RPE65 gene. Screening of her parents confirmed that both of these variations lie on separate alleles. (See the sequencing chromatograms in figures 5 and 6)
 |
 |
| click on image for enlarged image | |
Discussion: Leber congenital amaurosis typically presents with decreased vision during the first year of life with associated nystagmus. Vision can range from 20/30 to no light perception. Most patients with RPE65 mutations are profoundly night blind. Most patients also have sluggish pupils and hyperopia, although some patients such as this one are myopic. The fundus appearance varies widely from a retinitis pigmentosa-like appearance to retinal pigment epithelium changes, chorioretinal atrophy, macular coloboma, or a marbleized retinal appearance. Franceschetti s oculo-digital sign consists of eye poking, pressing, and rubbing to mechanically stimulate the retina to produce a sensation of light. Some patients have keratoconus caused by this frequent rubbing. The electroretinogram is severely reduced or non-recordable[1] in virtually all patients.
In a case-control study of 642 patients with clinically diagnosed LCA, the carrier frequency of the most common LCA allele, CEP290, was found to be 1/261. It was estimated that that this allele is responsible for 29.9% of LCA. Based on these numbers, a Hardy-Weinberg analysis estimated that the overall prevalence of LCA in the population is about 1/81,000[2]. This means approximately 3,000 people in the United States likely have LCA. It is caused by at least 15 genes; most of which are inherited in an autosomal recessive manner. CRX-associated LCA is an autosomal dominant disorder [3] but accounts for only a few percent of LCA.
In 2006, Wyc Grousbeck (the CEO and co-owner of the Boston Celtics) and Derrek Lee (1st baseman of the Chicago Cubs) teamed up with investigators at the University of Iowa to identify every man, female and child affected with LCA in the United States - about 3000 people and to make a genetic test for LCA available to them. This effort is called Project 3000 (www.project3000.org). It is estimated that nearly 85% of all children with LCA under the age of 10 have already been identified thanks to this effort.
In 2008, human gene therapy trials for RPE65-associated LCA were initiated. Most patients had some improvement in vision and there were no serious complications [4-6]. The greatest improvement was noted in children[7]. RPE65-associated LCA accounts for 8% of all LCA cases, which translates into approximately 4 new cases per year in the United States. A major goal of Project 3000 is to identify all individuals in the United States who may benefit from gene therapy as rapidly as possible.
Counseling Points:
- LCA is an autosomal recessive disorder (except for CRX-associated LCA)
- The residual risk of having another affected child for the parents of this child is 25%
- Pre-implantation genetic testing could be offered to the parents if they wished to have additional children.
- While some genetic subtypes of LCA have been associated with other systemic complications (renal failure, developmental delay and autism), RPE65-associated LCA seems to be isolated to the eye.
Summary: RPE65-associated Leber Congenital Amaurosis (LCA)
EPIDEMIOLOGY
|
SIGNS
|
SYMPTOMS
|
MANAGEMENT / COUNSELING POINTS
|
Differential Diagnosis:
- Early-onset retinitis pigmentosa
- Severe early childhood onset retinal dystrophy (SECORD)
- Achromatopsia
- Congenital stationary night blindness (CSNB)
- Intrauterine infection
- Autoimmune retinopathy
References
- Simon, J., Aaby, AA, Drack, AV, Hutchinson, A, Olitsky, SE, Plager, DA, Raab, EL, Morse, C, Edmond, J, Basic and Clinical Science Course: Pediatric Ophthalmology and Strabismus. Vol. 6. 2007, San Diego, CA: American Academy of Ophthalmology. 334-335.
- Stone, E.M., Leber congenital amaurosis - a model for efficient genetic testing of heterogeneous disorders: LXIV Edward Jackson Memorial Lecture. Am J Ophthalmol, 2007. 144(6):791-811.
- Drack, A.V., R. Johnston, and E.M. Stone, Which Leber congenital amaurosis patients are eligible for gene therapy trials? J AAPOS, 2009. 13(5):463-5.
- Maguire, A.M., F. Simonelli, E.A. Pierce, et al., Safety and efficacy of gene transfer for Leber's congenital amaurosis. N Engl J Med, 2008. 358(21):2240-8.
- Bainbridge, J.W., A.J. Smith, S.S. Barker, et al., Effect of gene therapy on visual function in Leber's congenital amaurosis. N Engl J Med, 2008. 358(21):2231-9.
- Hauswirth, W.W., T.S. Aleman, S. Kaushal, et al., Treatment of Leber congenital amaurosis due to RPE65 mutations by ocular subretinal injection of adeno-associated virus gene vector: short-term results of a phase I trial. Hum Gene Ther, 2008. 19(10):979-90.
- Maguire, A.M., K.A. High, A. Auricchio, et al., Age-dependent effects of RPE65 gene therapy for Leber's congenital amaurosis: a phase 1 dose-escalation trial. Lancet, 2009. 374(9701):1597-605.
suggested citation format:
Privett B, Stone EM. RPE65-associated Leber Congenital Amaurosis. February 16, 2010; Available from: http://www.EyeRounds.org/genetic/001-RPE65-LCA.htm.