
Blurry vision and headaches
A 44-year-old Vietnamese woman presented to the emergency department with a 10-day history of progressive blurry vision in both eyes and a three-day history of severe headaches. Her central vision loss had not improved with a refraction by her optometrist. Her severe occipital headaches worsened with movement and were associated with generalized malaise, extreme fatigue, mild photophobia, and tearing. Acetaminophen partially alleviated the pain.
She had recently traveled to Vietnam but denied encountering sick contacts there. She denied jaw claudication, fevers, or weight changes. She denied skin rashes, hearing changes, tinnitus, dizziness, numbness, or tingling. She denied ever having tuberculosis. She had no history of prior vision problems, autoimmune conditions, or cancer.
None
Acetaminophen as needed
No known drug allergies
No history of eye disease or autoimmune disease
She immigrated from Vietnam several years prior to presentation. She is married and has three children. She works at a nail salon. She does not consume tobacco products, alcohol, or illicit substances. She travels to Vietnam every six to twelve months.
Negative except for what is detailed in the history of present illness
Full extraocular movements in both eyes (OU)
Normal on both sides
 |
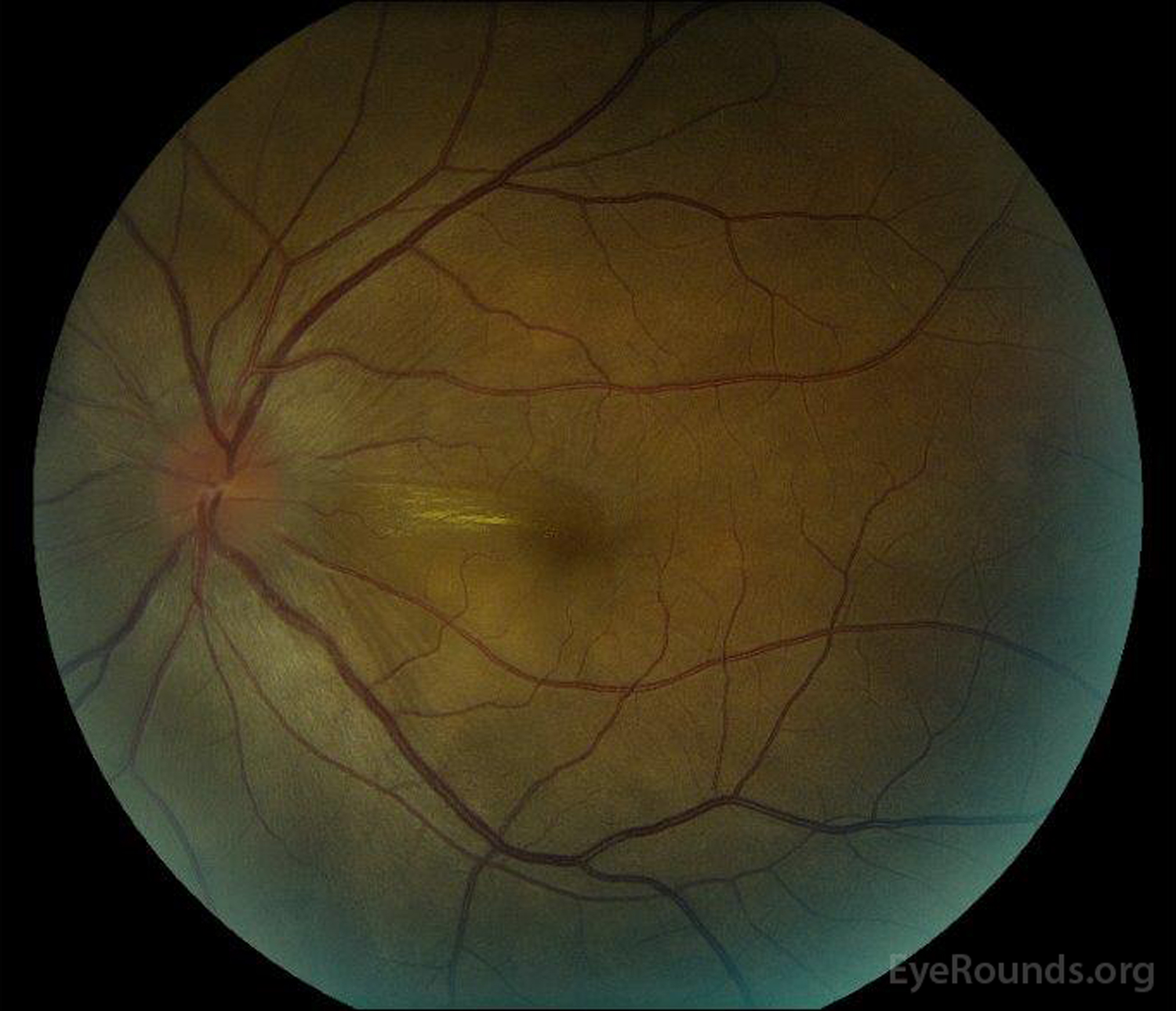 |
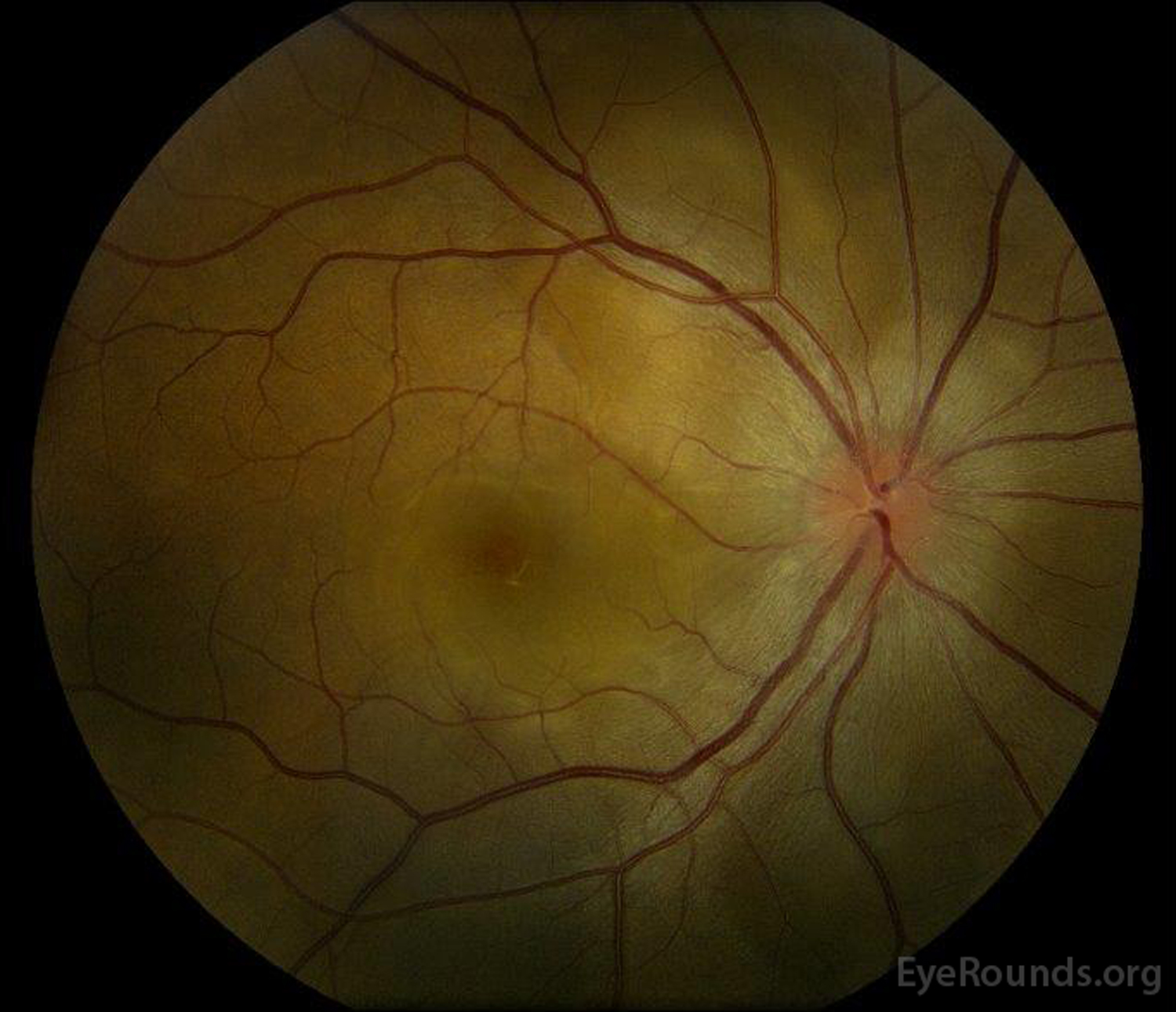
Figure 1: Color fundus photographs at presentation: (Left image) The right eye has disc edema and mild hyperemia as well as subretinal fluid extending from the disc temporally through the macula. There is also a focal serous retinal detachment superotemporal to the disc, along the superior arcade. (Right image) The left eye has disc edema and mild hyperemia, along with subretinal fluid extending from the disc to the macula and linear lipid deposits in the nasal macula.
 |
 |
 |
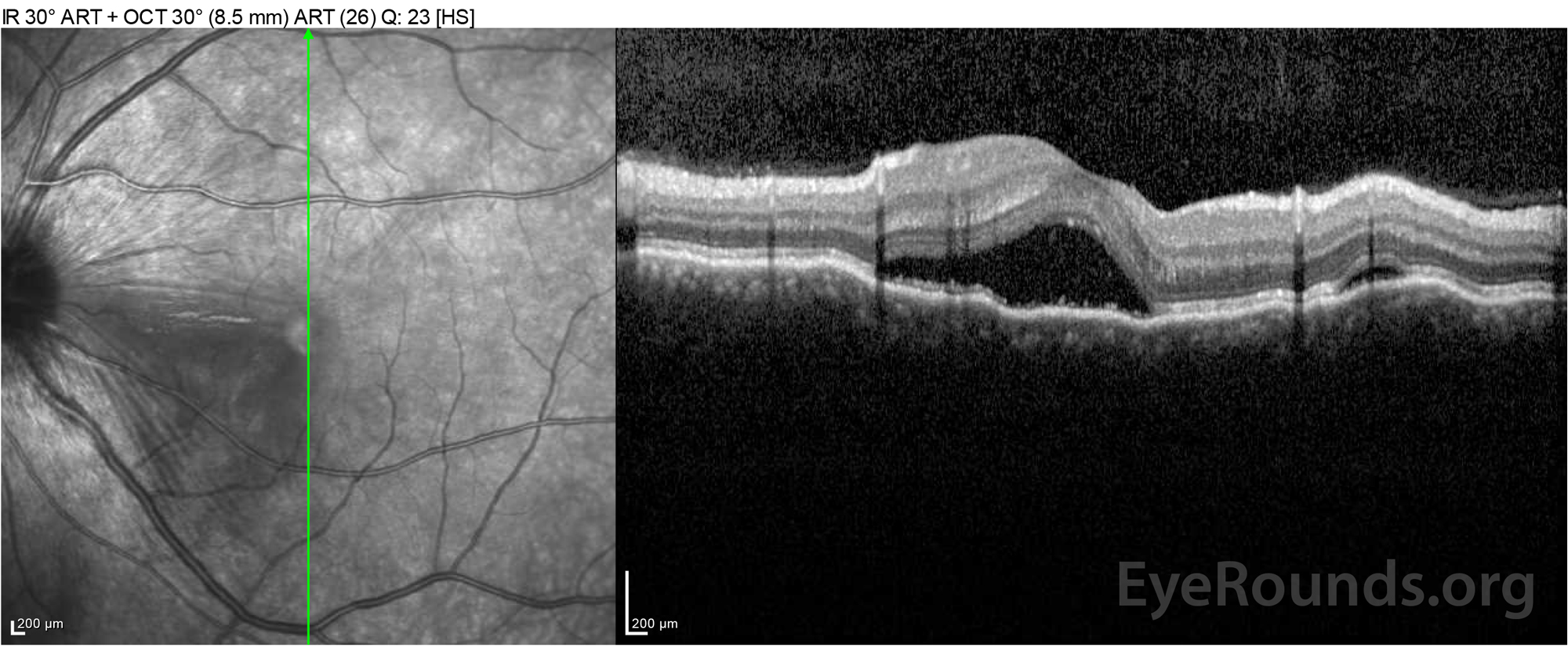 |
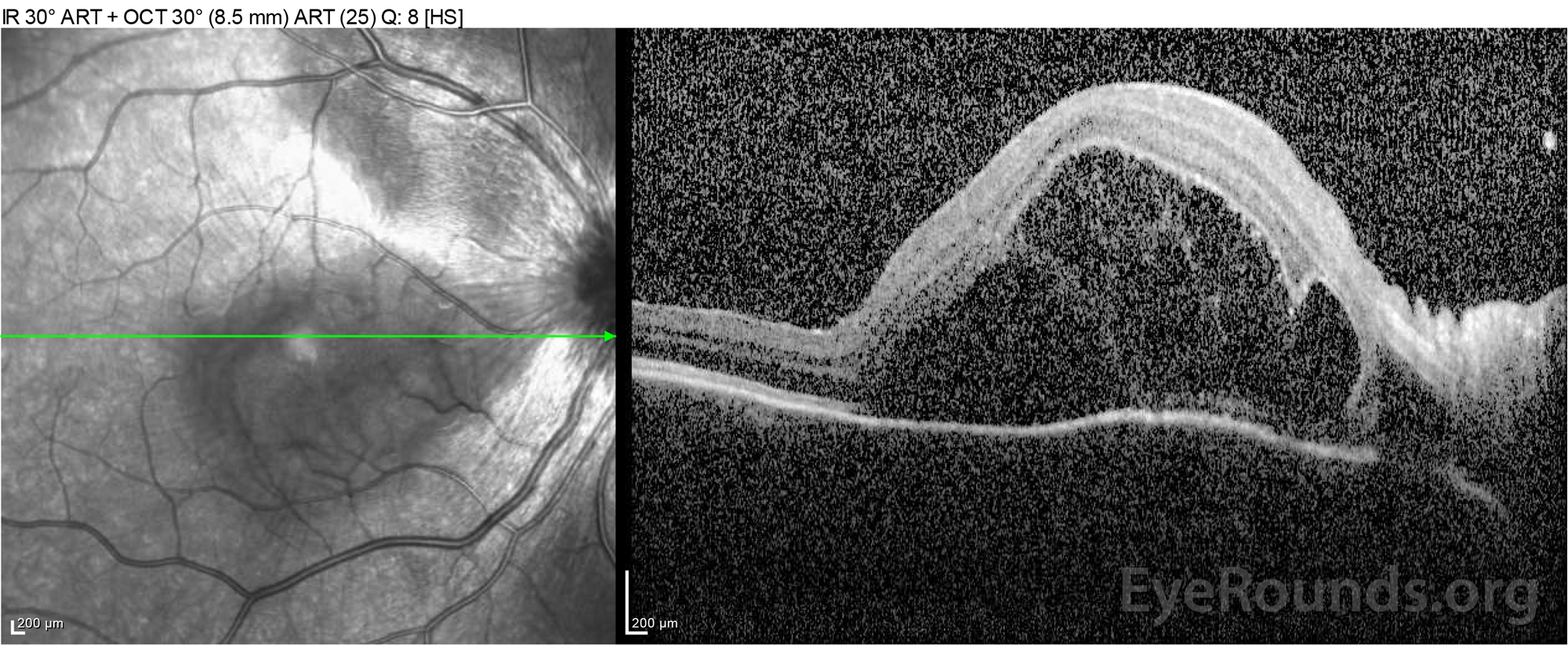
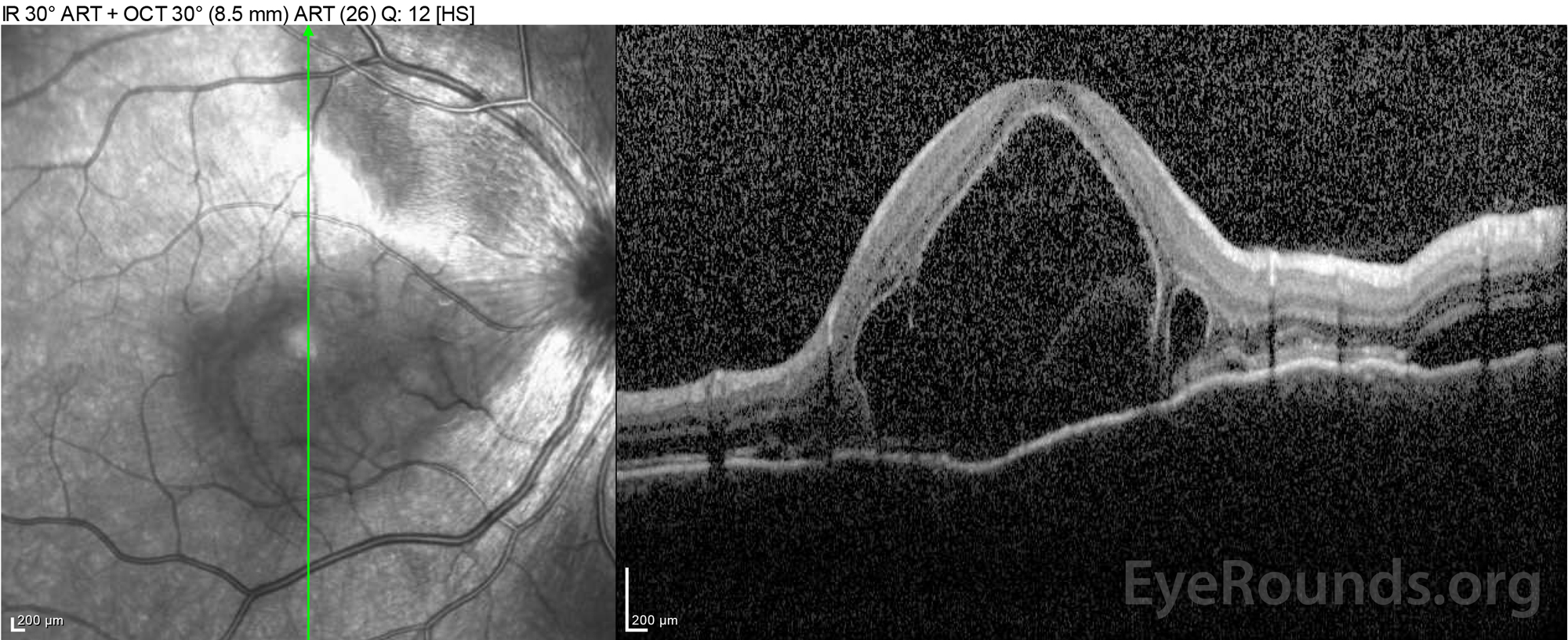
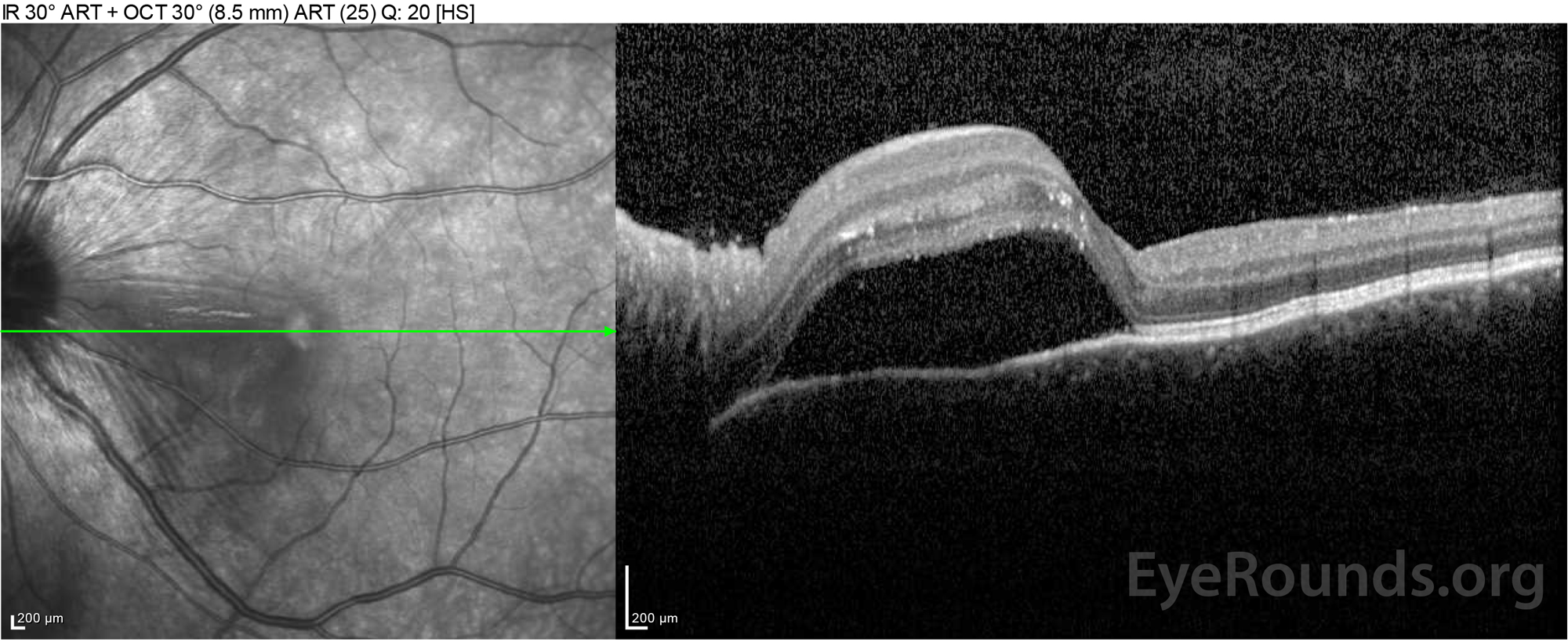
Figure 2: Optical coherence tomography (OCT) of the right eye (upper panels) shows a serous retinal detachment involving the fovea with extensive overlying intraretinal fluid, disruption of the outer retinal layers, and undulations of the thickened choroid. OCT of the left eye (bottom panels) shows a serous retinal detachment in the nasal macula extending up to the fovea.
 |
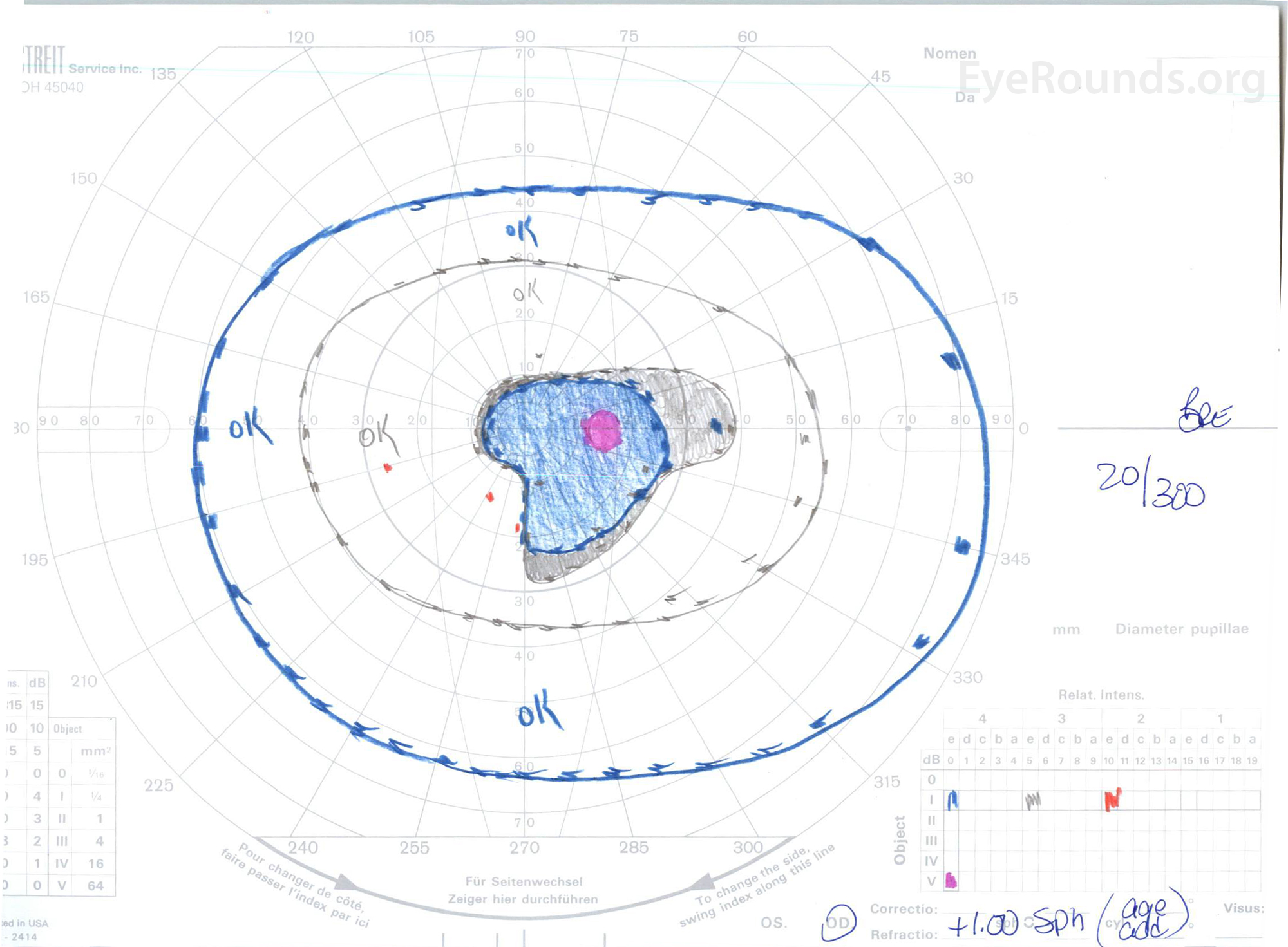 |
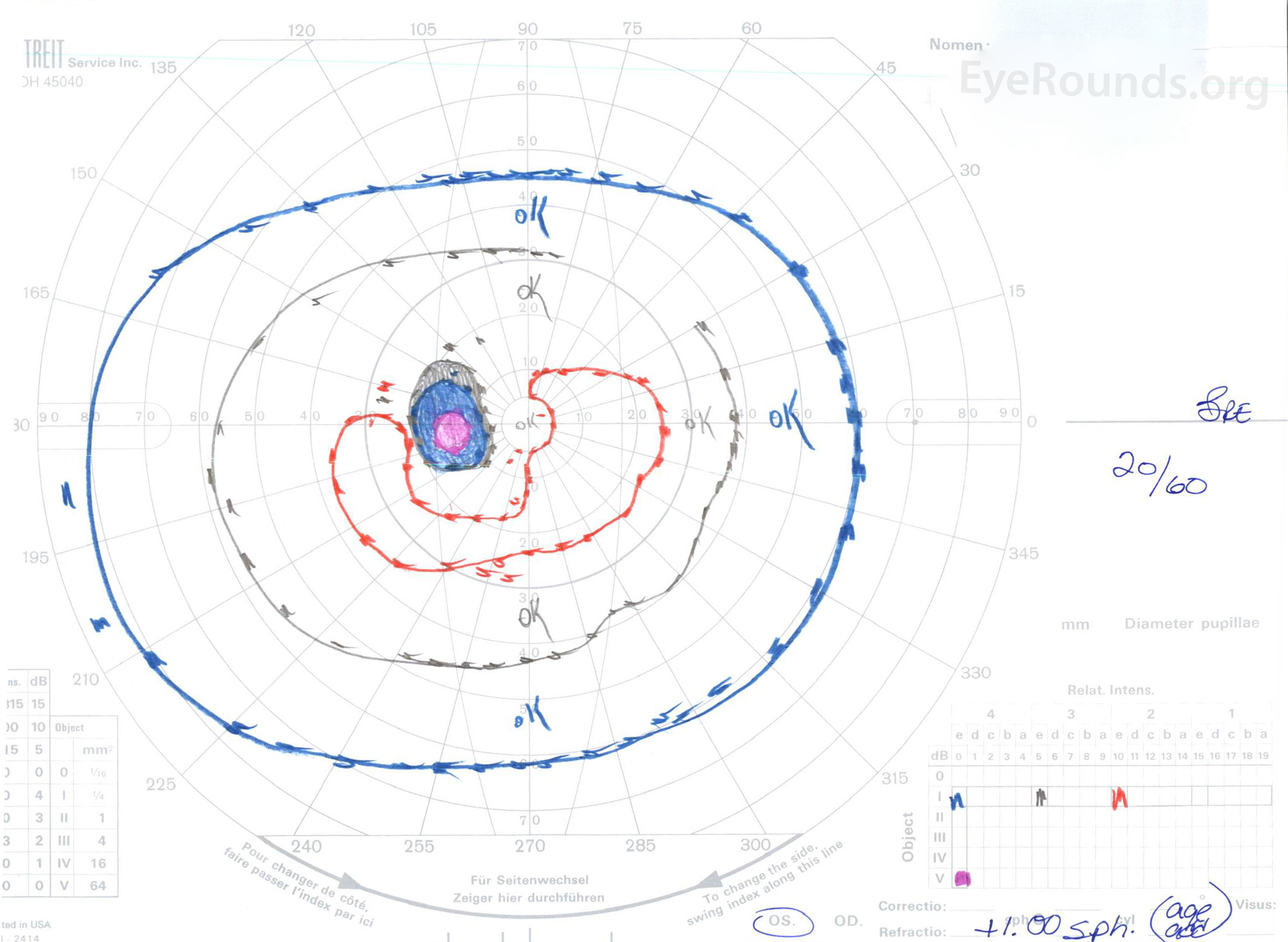
Figure 3: Goldman visual fields (GVF), OU. (Left image) OS shows an enlarged physiologic blind spot and mild central scotoma. (Right image) OD shows a moderately dense central scotoma incorporating the physiologic blind spot and extending inferotemporally.
B-scan: No signs of scleritis, mild vitreal opacities/cells inferiorly
Complete blood count
White blood cell count: 4.9 K/mm3 (Ref: 3.7-10.5)
Red blood cell count 3.99 M/mm3 (Ref: 4.0-5.2)
Hemoglobin 11.6 g/dL (Ref: 11.9-15.5)
Hematocrit 35 % (Ref: 35-47)
Basic metabolic panel
Sodium 138 mEq/L (Ref: 135-145)
Potassium 4.3 mEq/L (Ref: 3.5-5.0)
Chloride 107 mEq/L (Ref: 95-107)
CO2 20 mEq/L (Ref: 22-29)
Blood urea nitrogen 16 mEq/dL (Ref: 10-20)
Creatinine 0.7 mg/dL (Ref: 0.5-1.0)
C-reactive Protein (CRP): <0.5 mg/dL (Ref: <=0.5)
Erythrocyte sedimentation rate (ESR): 12 mm/Hr (Ref: 0-20)
Angiotensin--converting enzyme (ACE): 13 U/L (Ref: 8-52)
QuantiFERON-TB Gold: negative
Iron, blood 54 microgram/dL (Ref: 37-145)
Total iron binding capacity 379 microgram/dL (Ref: 250-425)
The patient was initially evaluated by the emergency department given her complaints of new-onset severe headaches and vision loss. Brain computed tomography (CT) and magnetic resonance imaging (MRI) scans were unremarkable. ESR and CRP were within normal levels. The ophthalmology clinic evaluated her the following day and found bilateral serous retinal detachments and panuveitis. ACE and QuantiFERON-TB Gold labs were both negative. She was diagnosed with Vogt-Koyanagi-Harada disease based on her clinical presentation and Asian descent. She was treated with 80mg of prednisone daily, acetaminophen as needed for headaches, and Vitamin D and calcium supplementation. Her headaches rapidly resolved, and her visual acuity steadily improved over the following two weeks. Her prednisone dosage was then tapered down to 40 mg over three weeks with continued resolution of symptoms and improvement in visual acuity. She had no recurrence of headaches or worsening vision during the prednisone taper. At her most recent appointment, she had tapered down to 5mg every other day, without return of symptoms. Her visual acuity at that follow-up visit was 20/15-2 OD and 20/20+2 OS, and macular OCT showed full resolution of disc edema and serous retinal detachments in both eyes (Figure 4).
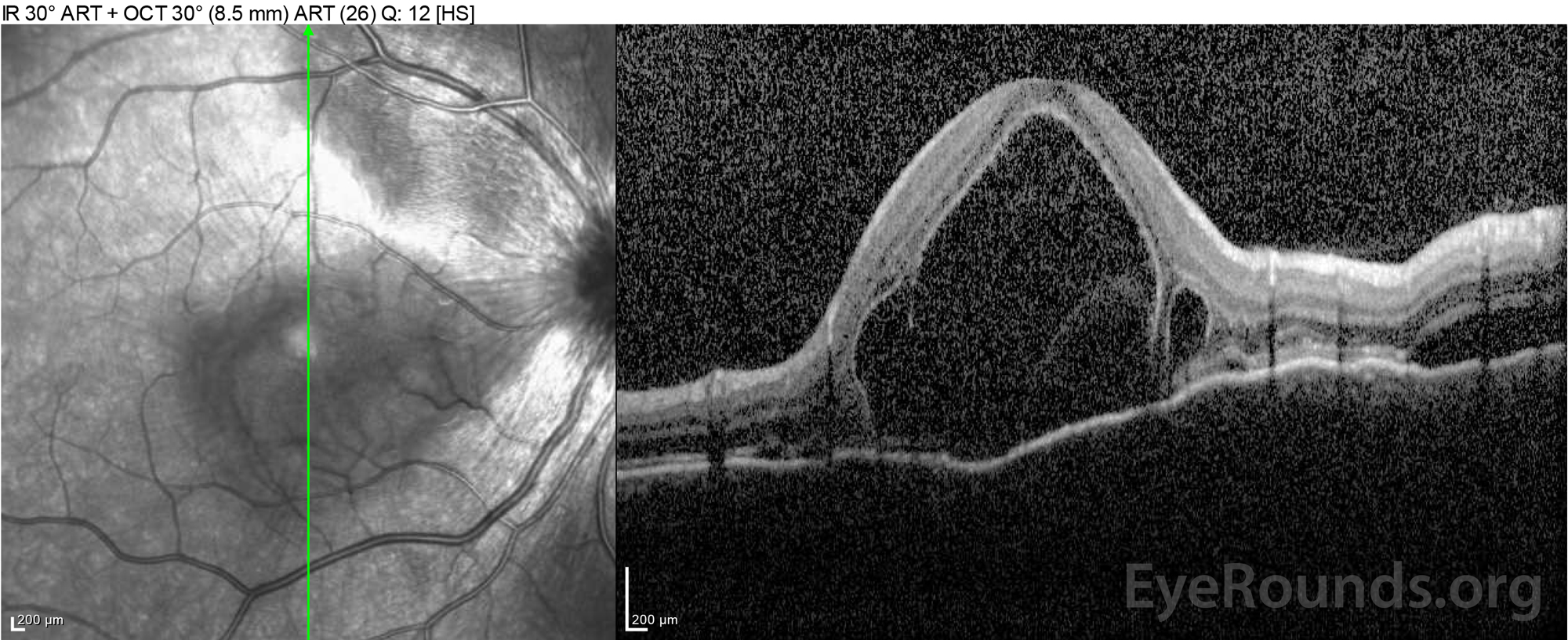 |
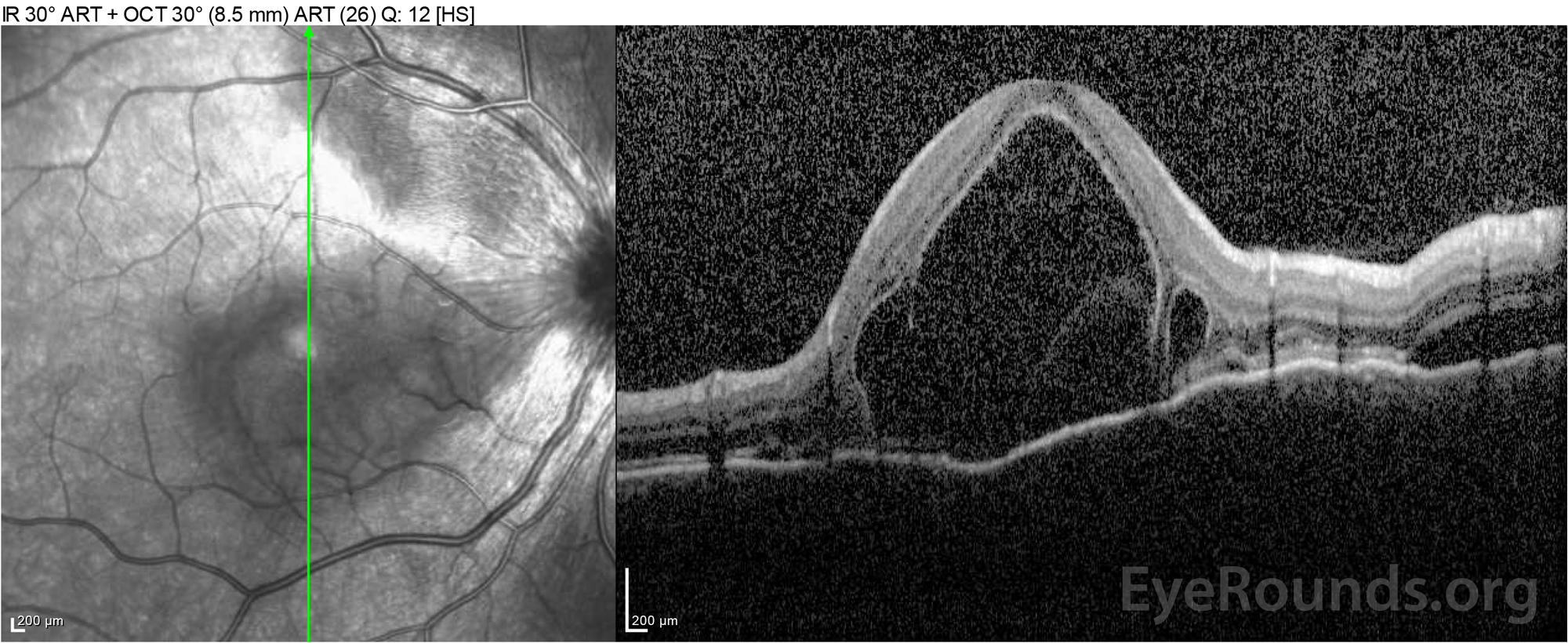 |
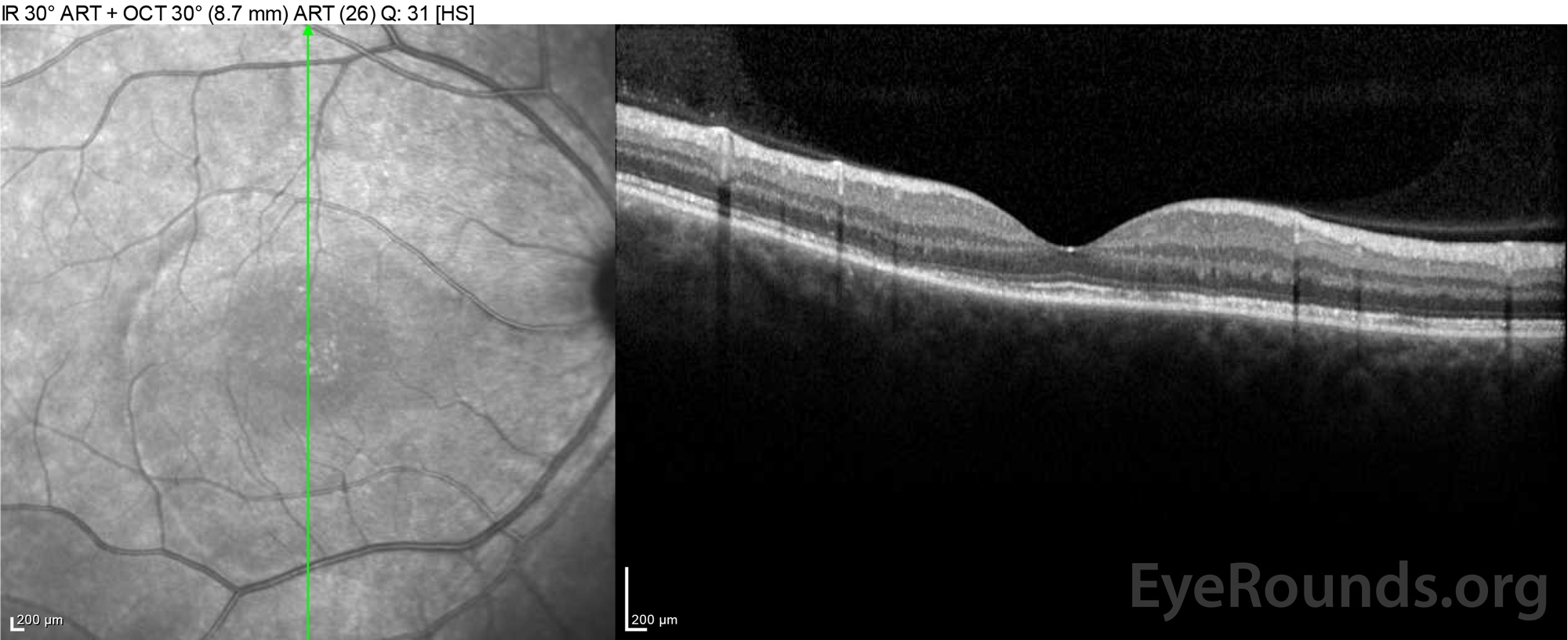 |
Figure 4: Optical coherence tomography showing subretinal fluid at baseline (top) and the course of resolution at one week (middle) and five weeks (bottom) while on a high-dose oral prednisone taper. Note the smoothing out of the choroidal undulations with treatment.
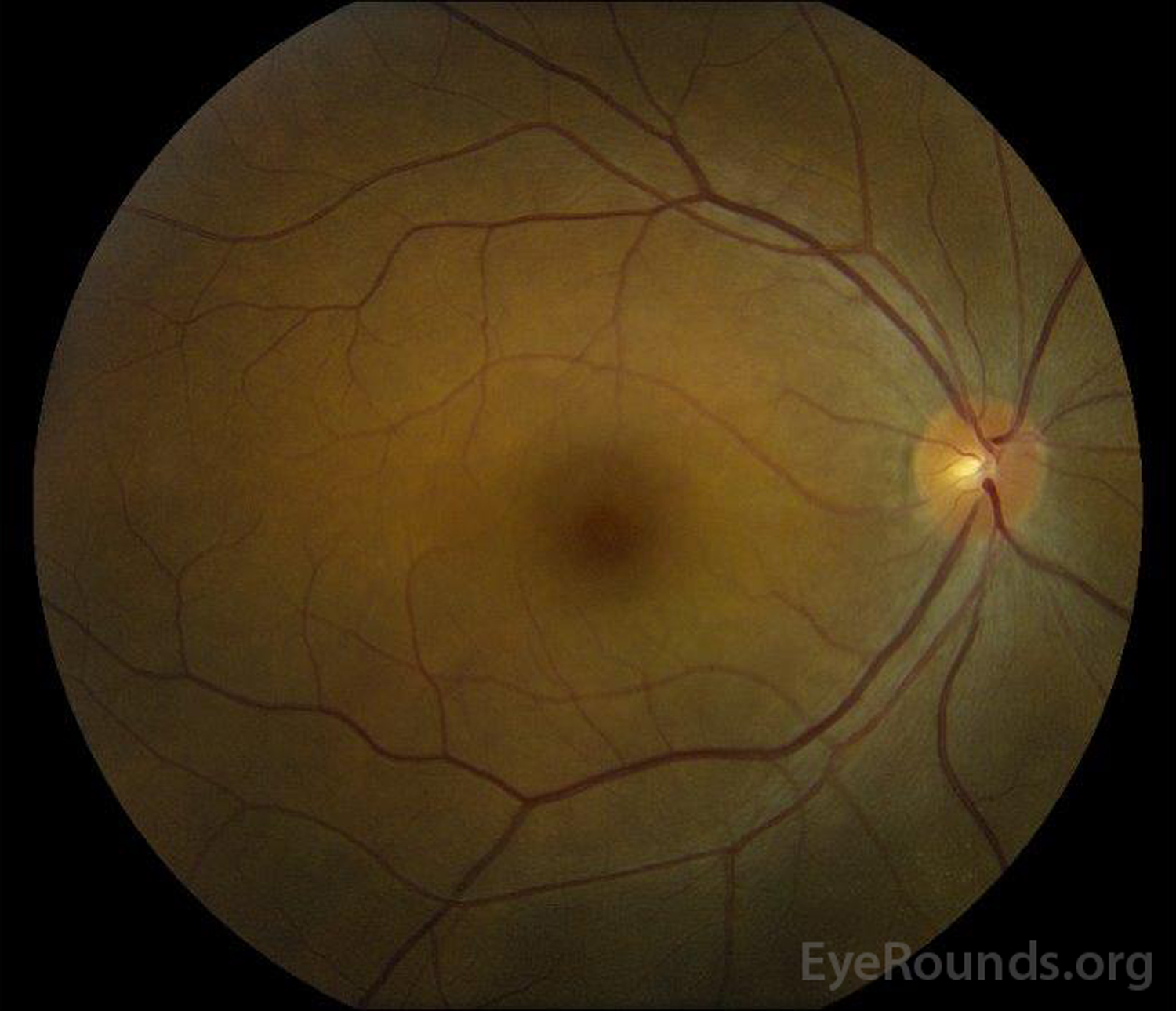 |
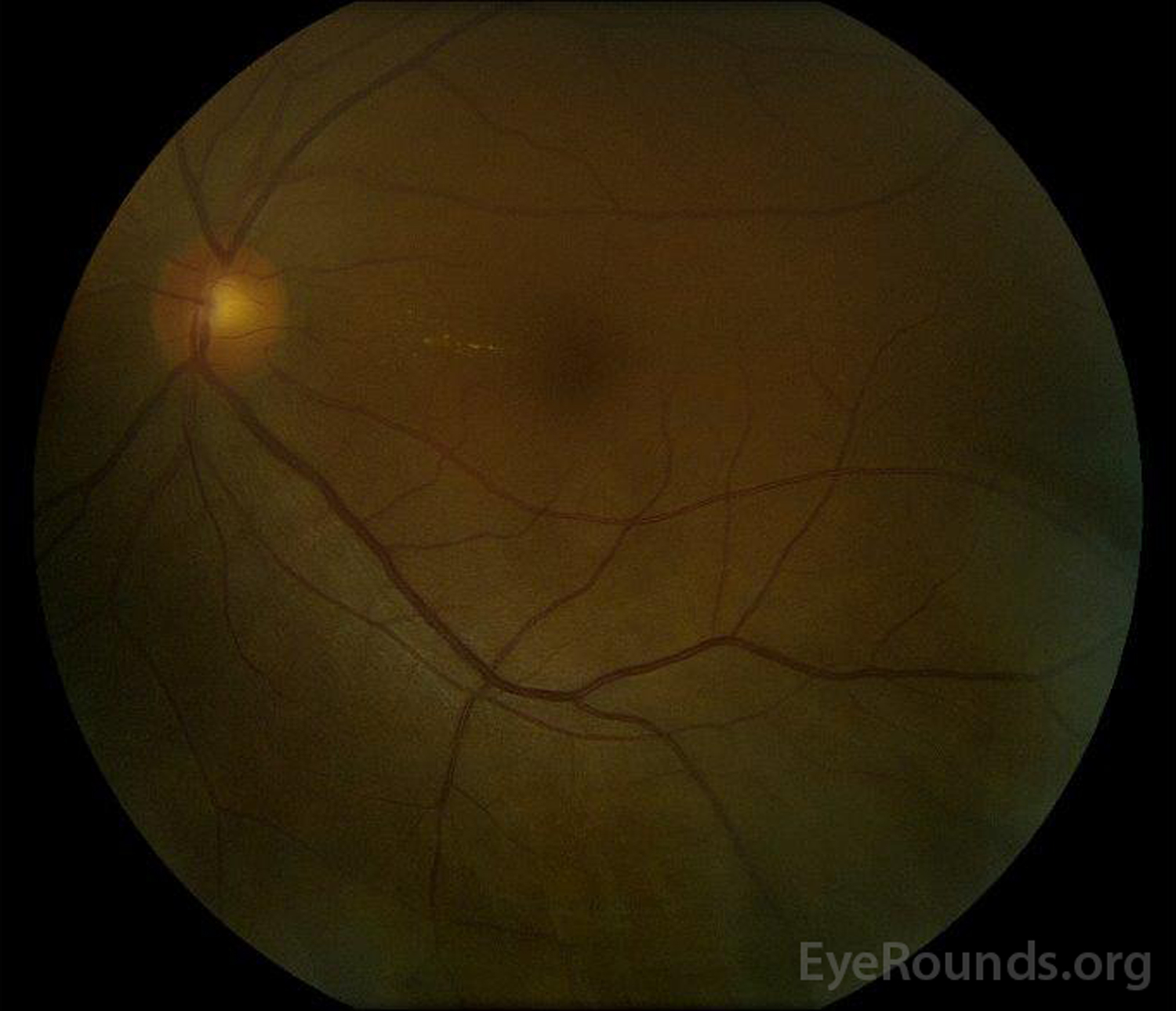 |
Figure 5: Color fundus photography of the right (A) and left (B) eyes during the convalescence phase demonstrating improvement in the subretinal fluid and disc edema.
Incomplete Vogt-Koyanagi-Harada Disease
Vogt-Koyanagi-Harada (VKH) Disease is a systemic autoimmune condition characterized by bilateral non-necrotizing granulomatous panuveitis associated with extraocular integumentary changes, such as poliosis and vitiligo, and inflammation affecting the uvea, inner ear, hair, and meninges. Harada disease is the isolated uveitis without the systemic signs or symptoms of VKH.
The etiology of VKH disease is still largely unknown despite current research efforts. It is thought to be an acquired autoimmune disease involving T-cell-mediated hypersensitivity to melanocytic self-antigens, with an underlying genetic predisposition and possible microbial trigger [1]. Tyrosinase and tyrosinase-related peptides are melanocyte antigens that have been suggested as targets of autoimmune processes in VKH [2]. However, increased risk of VKH disease was not associated with the tyrosinase gene family, according to one study [3].
Because of the increased prevalence among certain ethnic and gender groups, there is thought to be a genetic predisposition in the pathogenesis of VKH. Multiple genes, including human leukocyte antigen (HLA) and interleukin (IL) genes, have been associated with VKH in different ethnic populations [4]. HLA receptors are major histocompatibility complexes in humans that present peptides to the immune system. HLA-DR1, HLA-DR4, HLA-DRB1*0405, and HLA-DRw53 are several haplotypes found in patients with VKH disease [5,6]. HLA-DR4 is more common in Japanese and Hispanic people, while HLA-DRB1*0405 is more frequent in Korean and Middle Eastern patients [7]. Both the HLA-DR4 and HLA-DRB1*0405 alleles are found in Vietnamese patients [8]. The HLA-DRB1 receptor binds to melanocyte antigens in varying capacities. Despite these associations, genetic testing is not recommended at this time.
Given the usual prodromal symptoms that accompany VKH, including fever, headaches, meningismus and tinnitus, an inciting viral etiology has been suggested as a trigger for VKH onset through mechanisms of molecular mimicry in genetically predisposed patients. Cytomegalovirus envelope glycoprotein H has significant amino acid homology to the tyrosinase peptide, and CMV infection may trigger VKH through molecular mimicry (i.e., recognition by HLA-class II receptors) [9]. Ebstein-bar virus (EBV) has also been implicated. However, there has been no definitive evidence regarding a viral etiology of VKH, and it remains unclear what triggers the VKH autoimmune response [1].
There are four classic phases of VKH that can have variable presentations: prodromal, acute uveitic, convalescent, and chronic-recurrent. Histopathologic changes typically begin in the acute phase [7].
The acute uveitic phase is characterized by bilateral uveal thickening secondary to granulomatous inflammation. The granulomas consist of lymphocytes, macrophages, and granule-filled epithelioid and giant cells [10,11]. Although the epithelioid cells were previously believed to be altered melanocytes, a follow-up immunohistochemical study suggested an origin from tissue macrophages instead [1,12]. Granulomas filled with epithelioid histiocytes, termed Dalen-Fuchs nodules, can often be seen between the retinal pigment epithelium (RPE) and Bruch's membrane. The uveal granulomatous inflammation leads to choroidal thickening and exudative retinal detachments filled with proteinaceous fluid. In addition, pleocytosis (i.e., increased cell count) may be present in the anterior chamber and vitreous [7,11].
The convalescent phase is identified by depigmentation of the choroid and extraocular areas, including the periocular skin and hair. A depigmented choroid set against a pale optic nerve gives the impression of a "sunset-glow" fundus, which is a classic feature of this phase of VKH [10]. In addition, Dalen-Fuchs nodules become more prominent underneath the RPE in the convalescent phase [6].
The chronic-recurrent phase is characterized by decreased choroidal thickness, resolution of serous retinal detachments, chronic mild vitritis, and recurrent granulomatous anterior segment inflammation. Choroidal neovascularization (CNV) and subretinal fibrosis may develop during this phase and are indicators of severe disease progression [7,11]. Cataracts and secondary glaucoma are other complications of long-standing or recurrent inflammation in this phase [10].
VKH is prevalent in races with darker skin pigment, especially Asians, South Americans, Middle Easterners, and Native Americans. VKH disease accounts for >10% of uveitis in these populations [5-7,10,11,13,14]. Only 1-4% of uveitis cases are thought to be secondary to VKH disease in the United States (7). In the United States, most cases of VKH have been found to affect individuals of Asian, Hispanic, and/or Native American decent . Interestingly, VKH disease rarely affects Africans despite their dark pigmentation [15]. The incidence of VKH disease varies greatly between racial subgroups in neighboring countries [1,7,10] . For instance, Korea's incidence of VKH is only 2%, much lower than that found in Japan and China [16].
VKH has a typical onset of 20 to 50 years of age [17-20]; however, studies suggest that 3.1-13.4% of VKH cases are pediatric patients and 10% of cases are ≥65-years-old [1]. Classically, VKH is thought to have a predilection for the female gender, and while most studies show that VKH disproportionately affects women, a few studies have shown a male predisposition or no gender predisposition [21].
As aforementioned, the four stages of VKH disease are prodromal, uveitic, convalescent, and chronic recurrent. Each stage exhibits distinct clinical features.
The most recent diagnostic criteria, named the Revised Diagnostic Criteria (RDC) for VKH, were defined in 1999 at the First International Workshop on VKH [15]. These are outlined in Table 1. The RDC are useful in that they divide VKH into three different diagnostic categories based upon the disease phase during which a patient presents: complete, incomplete, and probable. This categorization of disease allows for appropriate and early management in "probable" disease that may help prevent progression to "complete" disease.
Work-up for other causes of ocular inflammation, both infectious and auto-inflammatory, are essential. These can include erythrocyte sedimentation rate (ESR), C-reactive protein (CRP), quantiferon-Gold testing for tuberculosis, rapid plasma reagin (RPR) for syphilis, angiotensin-converting enzyme (ACE) and a chest x-ray for sarcoidosis, antinuclear antibody (ANA), and p-/c-ANCA. Also, a history of recent ocular trauma or intraocular surgery must be noted and likely suggests sympathetic ophthalmia (SO) as the more likely diagnosis given the very similar presentation and pathophysiology shared between SO and VKH [25].
To support a diagnosis of VKH in equivocal cases, a lumbar puncture can be performed to look for lymphocytic and monocytic pleocytosis; however, this is rarely employed clinically. Eighty percent of patients have pleocytosis in the cerebrospinal fluid (CSF) within one week and 97% have pleocytosis within three weeks. Increased levels of immune cells can last up to eight weeks after disease onset [1,11] . The T-cell surface marker profiles are similar between the CSF and the aqueous humor but different from the blood. This suggests the ability of CSF to accurately reflect uveal inflammation in VKH disease [26].
*From Table 1 in (15).
"Complete Vogt-Koyanagi-Harada disease (criteria 1 to 5 must be present)
Incomplete Vogt-Koyanagi-Harada disease (criteria 1 to 3 and either 4 or 5 must be present)
Probable Vogt-Koyanagi-Harada disease (isolated ocular disease; criteria 1 to 3 must be present)
In the initial workup of VKH, one should consider obtaining the following tests:
Treatment goals in VKH include early diagnosis and suppression of active inflammation, along with prevention of recurrent inflammation and vision-threatening complications, such as glaucoma, bullous retinal detachment, and choroidal neovascularization.
Systemic corticosteroid treatment is the preferred therapy for VKH disease, especially during the acute uveitic stage. It has been shown that the route of corticosteroid administration (oral versus intravenous) does not impact the visual acuity or occurrence of visually-significant complications in the treatment of acute VKH [29]. For severe disease, the suggested protocol is intravenous methylprednisolone administration for three days followed by oral high-dose prednisone treatment. In mild-moderate disease, high-dose oral prednisone may be sufficient at 1-2mg/kg/day. The steroid dose should be slowly tapered over approximately six months to prevent recurrence [30]. Aggressive early treatment, alongside serial FA testing showing disappearance of dye leakage through the RPE, may help to prevent further disease progression, recurrence, and extraocular manifestations [7]. Topical steroids and cycloplegics can decrease cells in the anterior chamber and vitreous humor.
Intravitreal and sub-Tenon injections of triamcinolone have been used for short-term control of intraocular inflammation during the acute or recurrent phases; these local therapies should be considered in the case of recalcitrant disease and in patients who poorly tolerate the unfavorable systemic side effects of steroids given the extended steroid taper. Intravitreal anti-VEGF injections are sometimes used for control of choroidal neovascularization and in cases of persistent foveal serous retinal detachments [1, 5, 7, 25].
Steroid-sparing agents including antimetabolites, calcineurin inhibitors, biologics, TNF-alpha inhibitors, or cytotoxic agents can be used to treat VKH and should be carefully monitored, often in coordination with a rheumatology service [1]. There has been ongoing discussion regarding the use of non-steroidal immunosuppressive agents as first-line therapy for VKH disease. However, a recent study revealed no differences in outcomes between early first-line immunomodulatory treatment (IMT) and prednisone treatment alone [28]. Further, immunosuppressive and biologic therapies are expensive and require careful pre-treatment evaluation as well as frequent follow-up with blood work to assess for serious side effects.
In the chronic-recurrent stage, frequent recurrence may suggest resistance to corticosteroid therapy and suggests need for steroid-sparing immunomodulatory treatment [1, 6, 11]. The preferred agent for steroid-resistant recurrence or steroid-intolerance is cyclosporine [7]. Infliximab, rituximab, adalimumab, and interferon alpha-2a are biologic agents that have also been used to treat refractory uveitis in VKH disease.
To treat the anterior uveitis often associated with acute VKH, topical steroids (e.g., prednisolone acetate 1%) and topical cyloplegia (e.g., cyclopentolate 1% or atropine 1%) should be prescribed depending on the degree of anterior chamber inflammation.
Ocular complications are commonly associated with VKH disease. Given the multiple stages and variety of presentations in which a patient can present with VKH, treatment may be delayed in many cases. In severe forms of VKH and in recurrences, intraocular inflammation may be difficult to control and may result in structural damage. Over 50% of patients develop related complications, including cataract, secondary glaucoma, choroidal neovascular membranes, subretinal fibrosis, or a combination of these (Figure 6) [31].
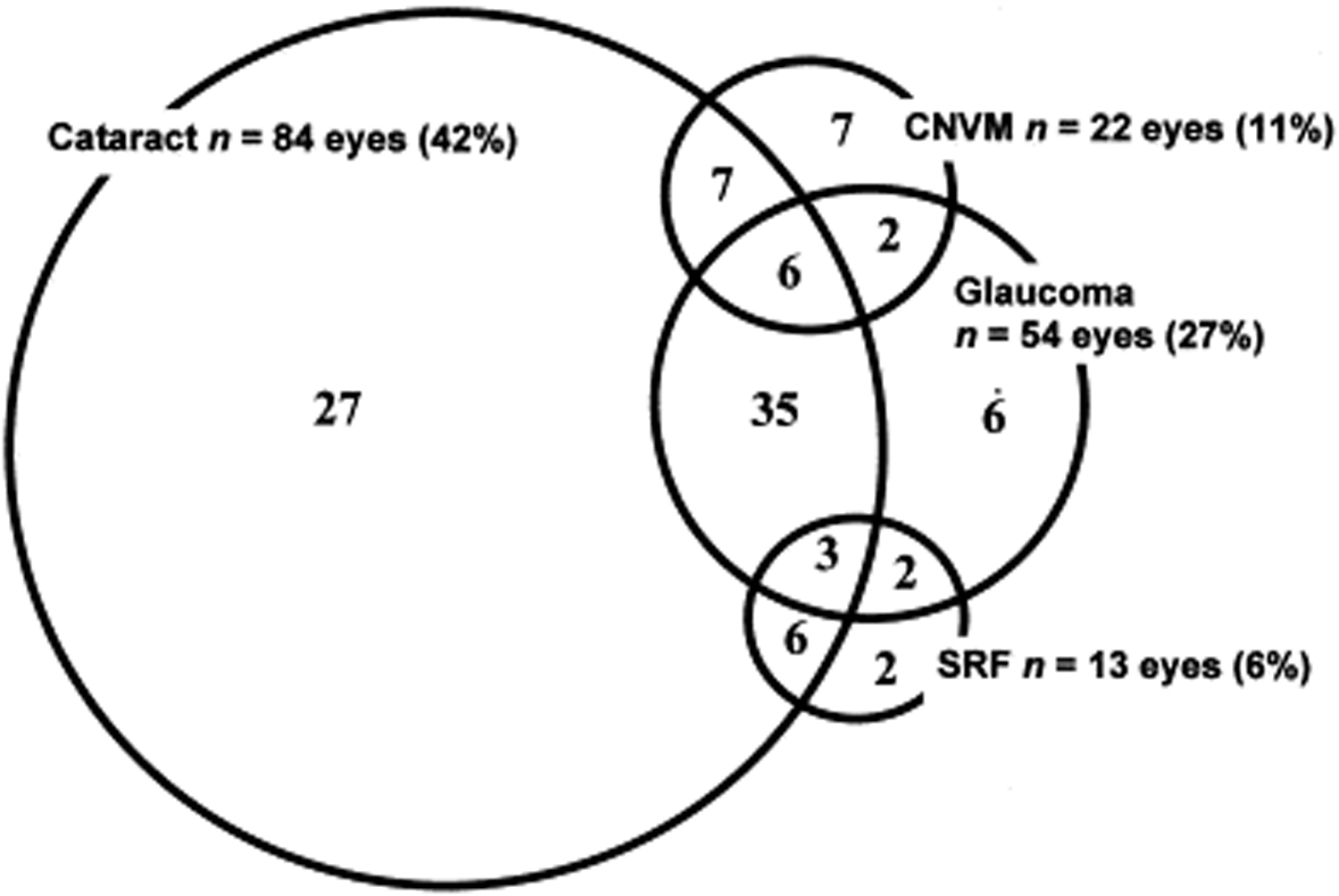
Figure 6: Venn diagram showing complications in VKH patients. (Used with permission from Am J Ophthalmol. 2001;131(5):599-606 [31])
EPIDEMIOLOGY AND ETIOLOGY
|
SIGNS
|
SYMPTOMS
|
TREATMENT/MANAGEMENT
|
Mai AP, Tran C, Wilson CW, Fox AR, Boldt HC. Vogt-Koyanagi-Harada (VKH) Disease. EyeRounds.org. April 1, 2019. Available from https://eyerounds.org/cases/284-vogt-koyanagi-harada.htm

Ophthalmic Atlas Images by EyeRounds.org, The University of Iowa are licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License.

Address
University of IowaLegal
Related Links
