
"The vision is blurry in my left eye."
A 69-year-old woman presented to the University of Iowa Hospitals and Clinics (UIHC) glaucoma clinic for evaluation of decreased vision in the left eye (OS) in the setting of elevated intraocular pressure (IOP) OS. Three weeks prior to her presentation at UIHC, she presented to her local optometrist for evaluation of blurring of her vision OS. At that visit, her IOP was 57 mmHg, and there was evidence of anterior chamber inflammation. Medical therapy was initiated with oral and topical aqueous suppressants, topical steroid drops, and a cycloplegic agent. Unfortunately, her IOP remained elevated; thus, she was referred to UIHC for further evaluation and management.
Penicillin G (hives)
No family history of glaucoma
Former smoker (quit 30 years prior)
Negative except for pertinent positives listed in HPI
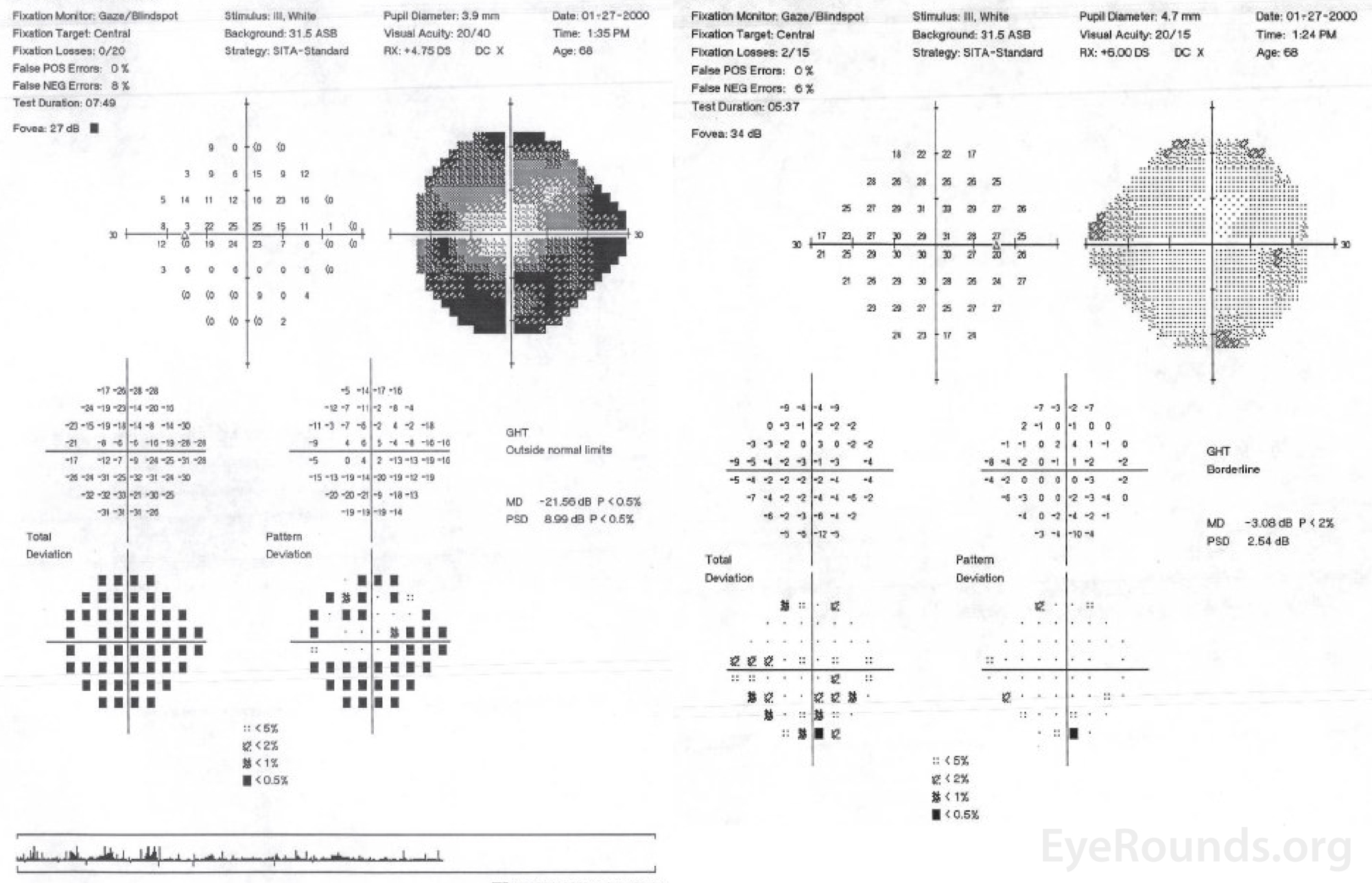
Figure 1. Central 24-2 Humphrey visual field, OU. OD (right HVF): Scattered non-specific depression, otherwise full. OS (left HVF): Dense superior and inferior arcuate loss with significant generalized depression.
At the initial visit at UIHC, brimonidine TID OS was added to the patient's drop regimen. Iridocorneal endothelial syndrome was suspected given the presence of unilateral glaucoma with iris nodules and signs of synechial angle closure. The decision was made to proceed with surgery given the persistently elevated IOP. Pre-operatively, the patient was given mannitol due to her severely elevated IOP. She underwent an uncomplicated trabeculectomy, and her intraocular pressure was 14mmHg on post-op day (POD) 1. She was discharged on prednisolone acetate 1%, atropine, and Tobradex ointment. At her appointment 10 weeks after surgery, visual acuity had improved to 20/30+2. She continued to have ectropion uveae, iris nevi, and pigment visible on the anterior capsule, but was stable with an IOP under 21 mmHg off all drops.
Three years after her initial trabeculectomy, the patient underwent phacoemulsification of a cataract in her left eye with intraocular lens placement at an outside institution. On POD1, she was noted to have an IOP of 27 mmHg; the IOP increased to 34 mmHg on POD3. She was started on brimonidine, latanoprost, and acetazolamide with subsequent fall of her IOP to 18 mmHg a week later. Her pressure subsequently spiked to 50 mmHg with discontinuation of acetazolamide. Acetazolamide was restarted, and timolol and prednisolone forte were added. The patient was referred back to UIHC, at which time her IOP was 20 mmHg OS, and she had a small relative afferent pupillary defect in the left eye. Visual acuity remained 20/20 in both eyes. Given her history of kidney stones, the acetazolamide was discontinued, and the patient was trialed on topical timolol-dorzolamide and oral acyclovir. Over the next two weeks, the patient's vision deteriorated. The IOP increased to 42 mmHg OS, and she developed corneal edema and bullae OS. Given her persistently elevated pressures and signs of corneal decompensation, the patient underwent a repeat trabeculectomy. This resulted in well-controlled IOPs off drops.
Over a year later, the patient was seen for a spike in her IOP to 38mmHg OS with accompanying blurred vision and pain OS. Humphrey visual fields showed substantial worsening from prior fields (Figure 2). Given uncontrolled IOP and worsening visual field deficits in the left eye, a Baerveldt seton implant was placed along with a surgical sector iridectomy to prevent any ICE-associated membranes from pulling the tube forward (Figure 3). On POD 1, her visual acuity was reduced to hand motions at 3 feet with an IOP of 30 mmHg OS and with a 1mm layered hyphema. Her visual acuity improved substantially over time, and at 6 weeks she was 20/30 with an IOP of 6mmHg OS. Two years later, her cornea showed further decompensation, which ultimately required a descemet stripping automated endothelial keratoplasty (DSAEK).
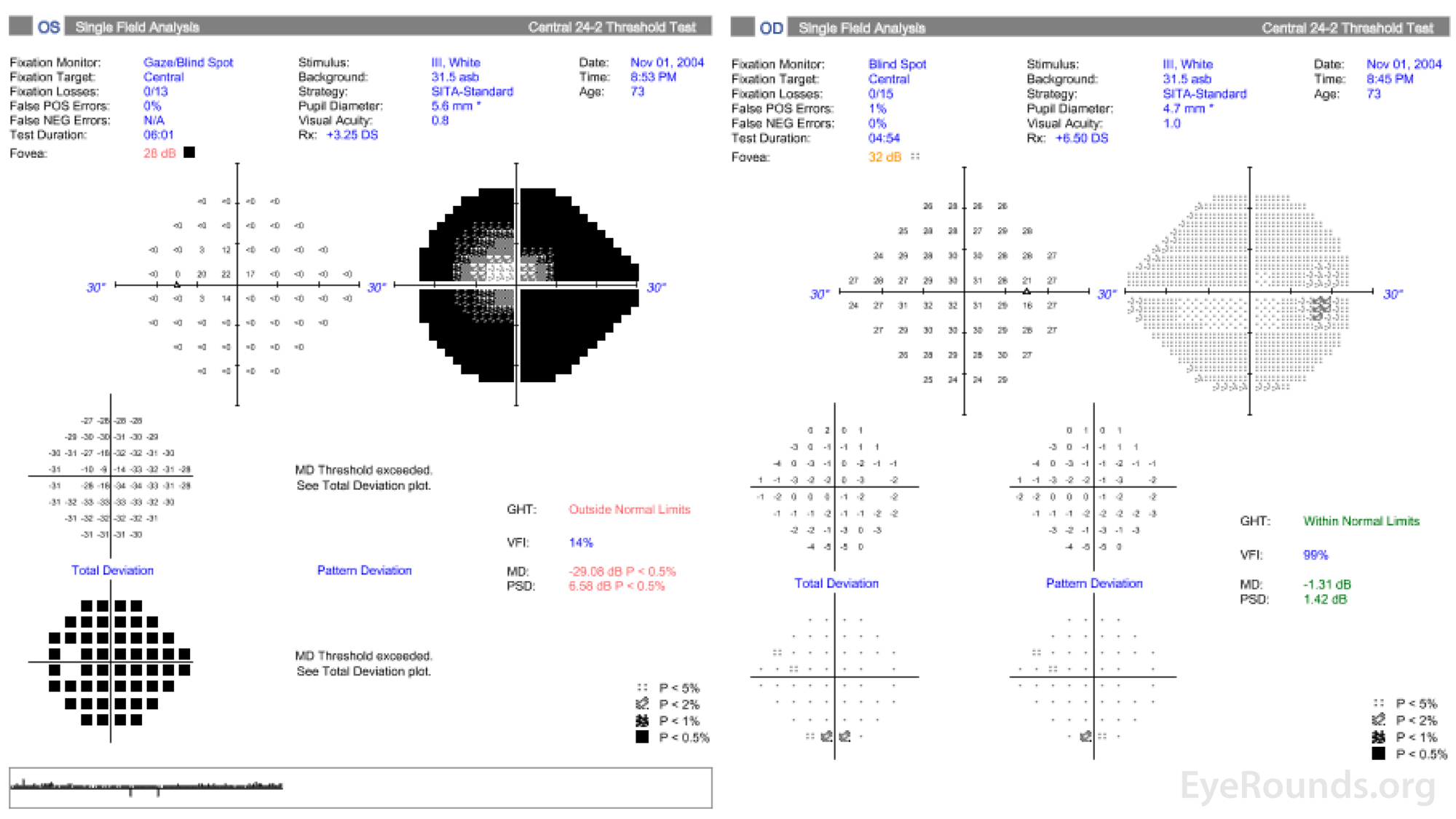
Figure 2. Central 24-2 Humphrey visual field, OU. OD (right HVF): Scattered non-specific depression, otherwise full. OS (left HVF): Dense superior and inferior arcuate loss with significant generalized depression. These visual fields represent a significant worsening OS despite having two prior trabeculectomies.
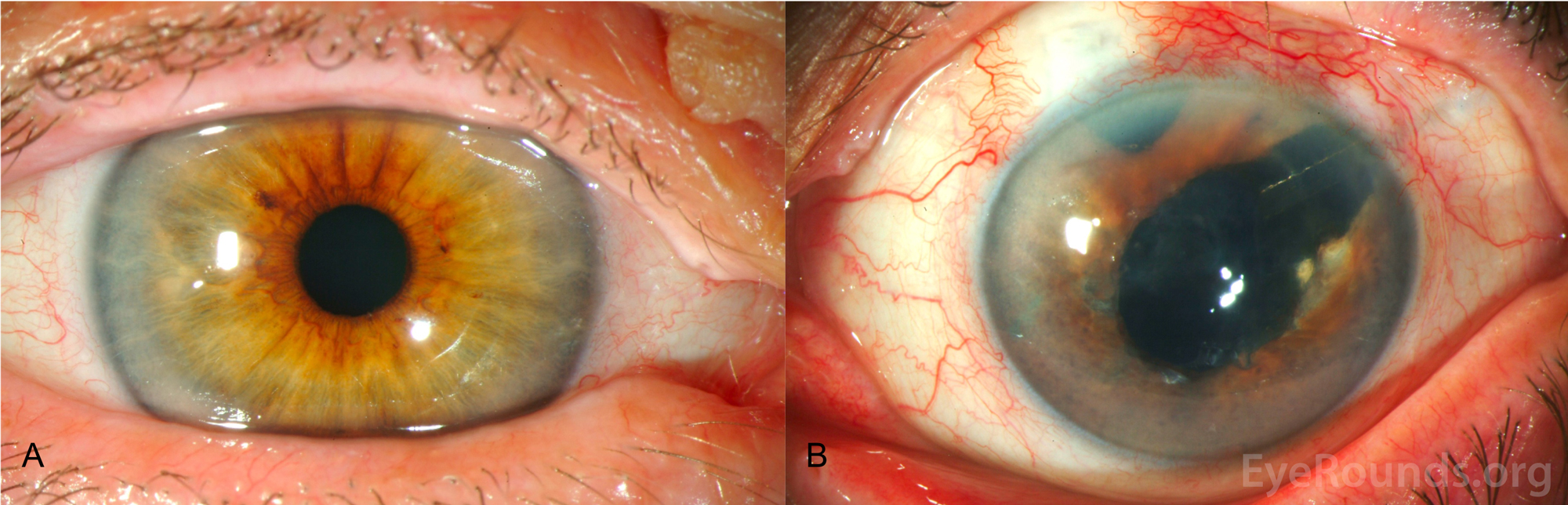
Figure 3. Slit lamp photography, OU. A: Slit lamp photo of the right eye shows no corneal or iris abnormalities. B: Slit lamp photo of the left eye shows corneal clouding secondary to diffuse stromal edema and bullous keratopathy. Multiple fine nodules distributed over the inferior 180 degrees; see Figure 4 for higher magnification of these nodules. Superotemporal Baerveldt seton with underlying surgical sector iridectomy in addition to avascular blebs superiorly and temporally from prior trabeculectomies. The patient was pseudophakic OU.
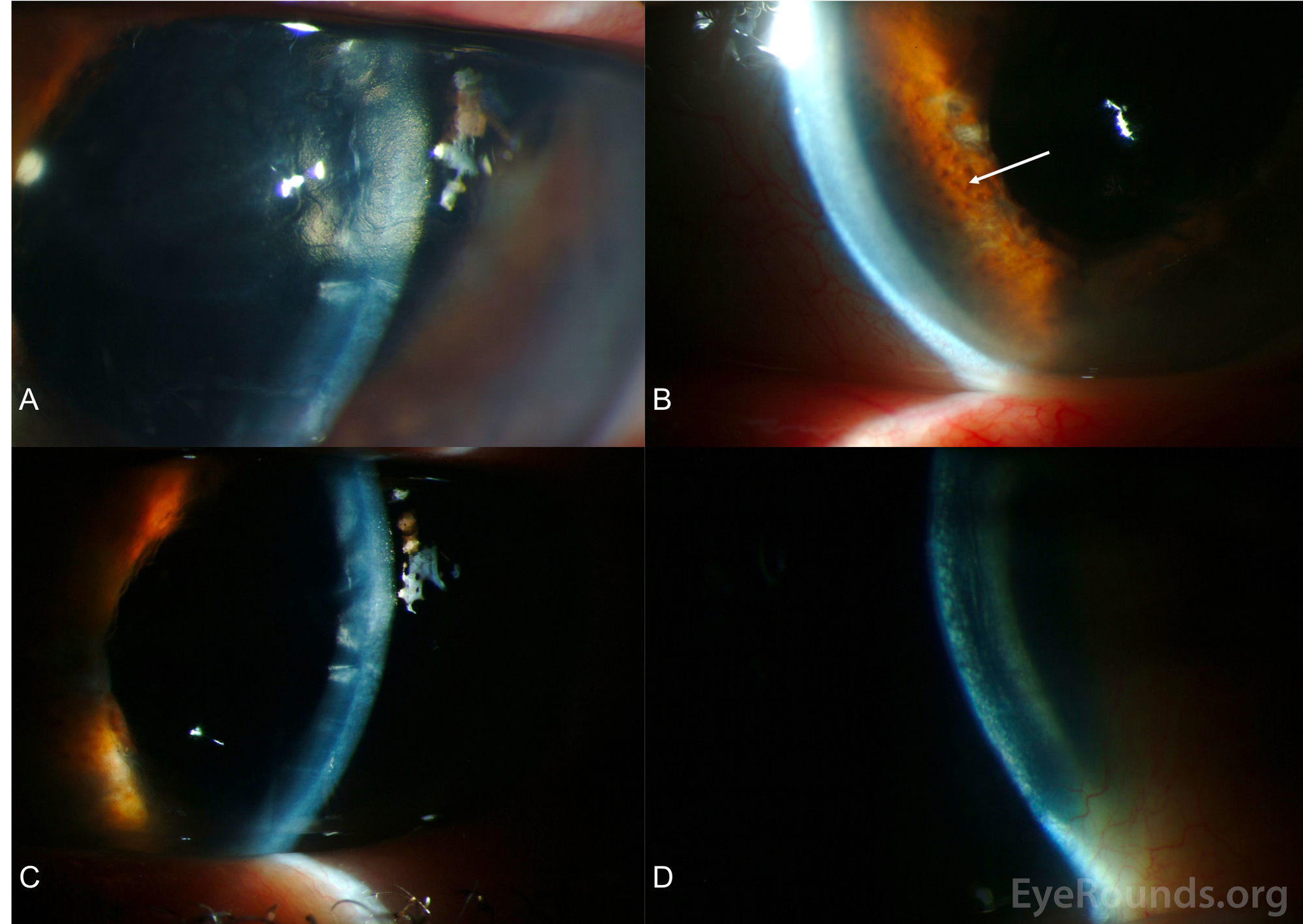
Figure 4. Slit lamp photography, OS. A. The patient had a "beaten metal" appearance of the corneal endothelium, which is classically seen in ICE syndromes. B. Consistent with Cogan-Reese syndrome, there were multiple fine nodules (arrow) distributed over the inferior 180 degrees of the iris. C. There were prominent descemet's folds with diffuse epitheliopathy. D. There was 2+ stromal edema with multiple bullae and 3+ microcystic edema throughout cornea. The patient had further corneal decompensation and underwent descemet stripping automated endothelial keratoplasty 1 month after these photos were taken.
Cogan-Reese Syndrome
Iridocorneal endothelial (ICE) syndrome is characterized by unilateral abnormal proliferation of the corneal endothelium, corneal edema, and peripheral anterior synechiae (PAS). Cogan-Reese syndrome, also known as iris nevus syndrome, is a very rare disease that is considered one of the three variations of ICE syndrome [1]; the other two ICE variants are Chandler's syndrome and Essential Iris Atrophy. All three disease entities are difficult to treat given their progressive nature and the obliteration of the iridocorneal angle that leads to refractory elevation in IOP. Similar to our patient, many affected individuals require multiple glaucoma procedures to normalize IOP, in addition to corneal endothelial keratoplasty (EK) to address the significant corneal decompensation.
Cogan-Reese syndrome most often presents between the ages of 20 and 50 years, and it is more common in women. All ICE syndromes are sporadic with no known family history of similar processes. Cogan-Reese is estimated to account for fewer than 10% of all ICE cases [2] with progressive iris atrophy and Chandler syndrome making up the majority of cases. ICE syndrome is unilateral in approximately >90% of affected patients [2], and the ICE variants are not associated with systemic disease.
The definitive mechanism of Cogan-Reese syndrome is unknown. However, polymerase chain reaction (PCR) analysis of the abnormal corneal endothelium in Cogan-Reese patients has demonstrated viral DNA. Herpes Simplex Virus has been found in a substantial number of patients with ICE syndrome yet is absent in normal corneal endothelium [3]. The proposed mechanism for the endothelium alteration in ICE syndrome is a shift in phenotype towards epithelial cells [4]. The dysregulation and metaplasia of the corneal endothelial cells are likely due to the downregulation of cell cycle regulators, specifically cyclin dependent kinases, in the corneal endothelium [5]. The abnormal endothelial cells in ICE syndrome are thought to have a toxic effect on neighboring healthy cells, thus explaining the progressive nature of the disease [6]. These abnormal endothelial cells migrate and ultimately obstruct the trabecular meshwork, thus causing high PAS and/or secondary angle-closure glaucoma; they also proliferate along the iris leading to iris thickening and color changes (Figure 4B).
The corneal endothelium in ICE syndrome is typically described as "hammered silver" or "beaten bronze" in appearance when viewed through the slit lamp using specular reflection (Figure 4A) [7]. Secondary, unilateral angle-closure glaucoma can occur in these patients because of the occlusion of the trabecular meshwork by the abnormal endothelial proliferation or PAS [4]. Cogan-Reese syndrome is unique among the ICE syndromes due to the presence of pedunculated, pigmented iris nodules, commonly referred to as iris nevi [1]. The classic Cogan-Reese nodules, which result from melanocytic proliferation within the iris stroma, are present on the anterior surface of the iris, appear late in the disease, and become darker and larger over time [6]. While these nodules are typically described as diffuse, it is possible for only a segment of the iris to be affected. Loss of the normal iris crypts leads to a smooth-appearing iris. Additionally, eversion of the pupil (i.e., ectropion uveae) and heterochromia may be present [8].
Making the diagnosis of an ICE syndrome is often challenging, as it can appear similar to other disease states, including posterior polymorphous corneal dystrophy. However, prompt diagnosis is important given the progressive nature and aggressive course of ICE. Specular microscopy is often helpful given the presence of "ICE cells" is pathognomonic. These cells are large, dark areas with a light center and light peripheral zone [6]. Histologically, these endothelial cells have epithelial features, such as microvilli and desmosomes [9]. In vivo confocal microscopy can also be used to detect the abnormal epithelioid-like endothelium. Gonioscopy of our patient was performed to better evaluate the iris and angle to determine the extent of secondary angle closure (Video 1). However, corneal edema often limits the view of the angle in Cogan-Reese syndrome, and such cases may benefit from the use of anterior segment optical coherence tomography (OCT) or ultrasound biomicroscopy to detect PAS or iridocorneal abnormalities [6]. Specific findings on anterior segment OCT that aid in detecting Cogan-Reese syndrome include increased iris thickness [10]. There may be utility in performing HSV PCR given this virus may contribute to the pathogenesis of the disease.
Management of Cogan-Reese, similar to that of the other ICE syndromes, is aimed at slowing or preventing vision loss from secondary angle-closure glaucoma by lowering the patient's intraocular pressure. Although herpetic disease may contribute to the pathophysiology of ICE syndrome, there are no data showing the benefit of antivirals for the treatment of ICE [6]. Medical management involves the use of aqueous suppressants. Unfortunately, this often fails to adequately control intraocular pressure due to progressive obstruction of the trabecular meshwork. A study of 200+ patients with ICE suggested that more than 50% of these patients require surgical intervention to control IOP [2]. Specifically, if medical management fails, a trabeculectomy or implanting a glaucoma drainage implant (GDI) should be pursued. Trabeculectomy is commonly performed, but the success rates are poor and many patients (12.5% to 53.8%) subsequently require a GDI [6, 7, 11]. The long-term outcome of the surgical treatment in ICE syndrome patients has been shown to be better in those with a GDI compared to a trabeculectomy [12], and this is likely due to the robust, post-trabeculectomy inflammatory response and the ICE cohort being relatively young [6]. Unfortunately, even surgical placement of a GDI leads to failure in approximately 20% of ICE patients; this is usually related to the filtering ostium becoming obstructed by continued growth of the proliferating endothelial membrane [6]. A sector iridectomy, which was performed at the time of GDI in our patient, should be considered in order to eliminate the tube being pulled or clogged by ICE-related membranes.
Corneal clarity is often compromised in Cogan-Reese patients. Hypertonic saline can be implemented in mild cases of corneal edema. However, if corneal decompensation is severe, endothelial keratoplasty (EK) or penetrating keratoplasty (PK) is required. A shallow anterior chamber, extensive PAS formation, and iris abnormalities may make graft positioning challenging for the EK surgeon [6]. It is estimated that 15% of patients with ICE syndrome will require keratoplasty [2], and a significant number of patients undergo repeat grafts due to high keratoplasty failure rates [13]. Comparing DSAEK vs. PK in ICE patients, two studies reported the rejection rates to be 33% vs. 79% [14, 15]; this suggests that EK surgery may be the favored procedure in Cogan-Reese. Several studies have demonstrated that pre-existing glaucoma surgery and ICE syndrome are both independent risk factors for descemet's stripping endothelial keratoplasty failure [14, 16, 17].
Cogan-Reese Syndrome is a rare iridocorneal endothelial syndrome that most commonly affects young- to middle-aged women. The patient may present with unilateral reduced vision, noticeable color changes to the iris, or unilateral secondary glaucoma due to corneal endothelium obstruction of the trabecular meshwork. The Cogan-Reese variant of the ICE syndromes is unique because of the pedunculated, pigmented nevi that present on the iris, in addition to the corneal edema, PAS, and ectropion uveae. The early identification of unilateral glaucoma should prompt a focused assessment for ICE syndrome, despite its unknown prevalence.
EPIDEMIOLOGY OR ETIOLOGY
|
SIGNS
|
SYMPTOMS
|
TREATMENT/MANAGEMENT
|
Madhan N, Scruggs BA, Carter PC, Melendrez J, Bettis, DI. Cogan-Reese Syndrome: A Rare Case of Unilateral Glaucoma. EyeRounds.org. posted July 20, 2018; Available from https://EyeRounds.org/cases/275-cogan-reese-syndrome.htmCitation

Ophthalmic Atlas Images by EyeRounds.org, The University of Iowa are licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License.

Address
University of IowaLegal
Related Links
