
Worsening binocular diplopia
A 45-year-old woman with an unremarkable medical history developed sudden painless onset of horizontal diplopia while driving and noticed her right eye (OD) turning inwards. She was evaluated locally by a neurologist, and an MRI brain and orbits with and without contrast was ordered and initially read as normal. The diplopia was thought to be secondary to a microvascular sixth nerve palsy OD. She was lost to follow-up and later presented to the University of Iowa Hospitals and Clinics (UIHC) one year after onset of symptoms. At presentation to UIHC, she reported constant horizontal binocular diplopia in right gaze more than left gaze. She did not have diplopia with her preferred right head turn. She denied any trauma, headaches, eye pain, vision loss, pulsatile tinnitus, proptosis, periorbital edema, or eye redness.
None
None
Daily multivitamin
No known drug allergies
Non-contributory
The patient moved to Iowa from Vietnam 5 years prior to presentation, and she worked in a restaurant. She denied tobacco or alcohol use.
Energy level was reported as being 70% of normal. She reported occasional numbness of toes and fingers and muscle cramps of her legs.
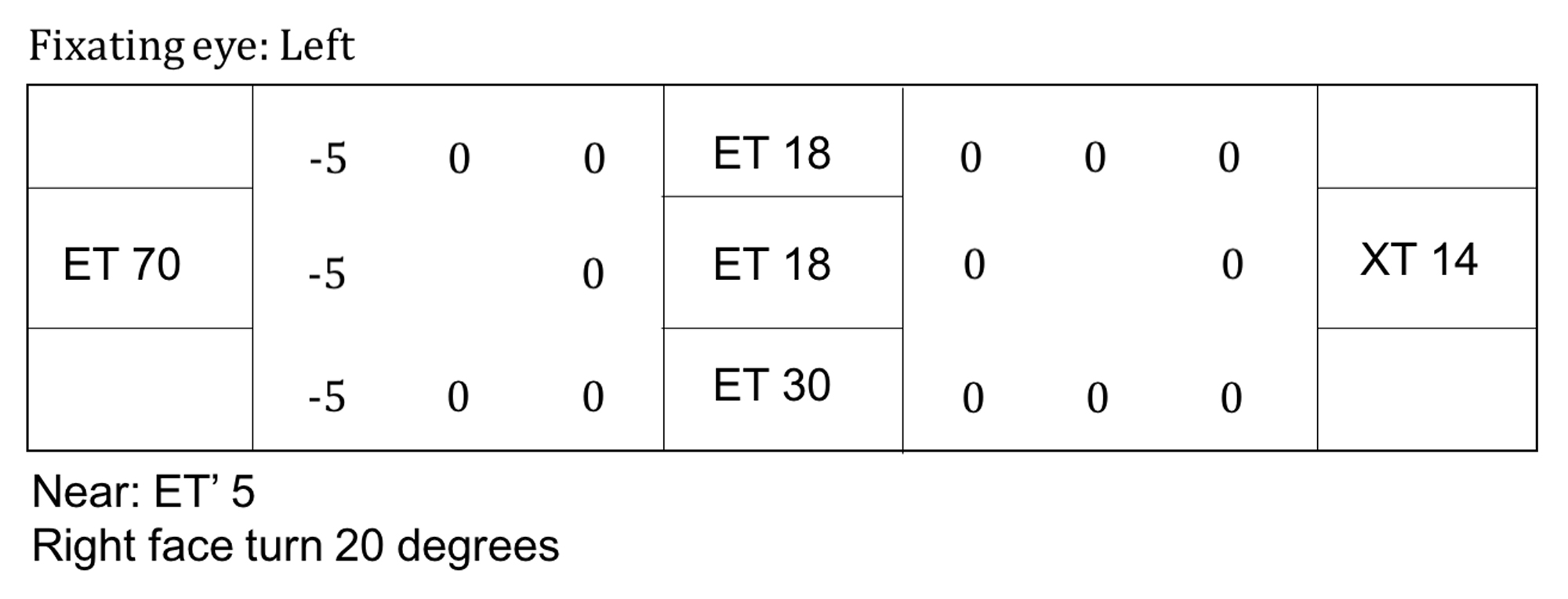
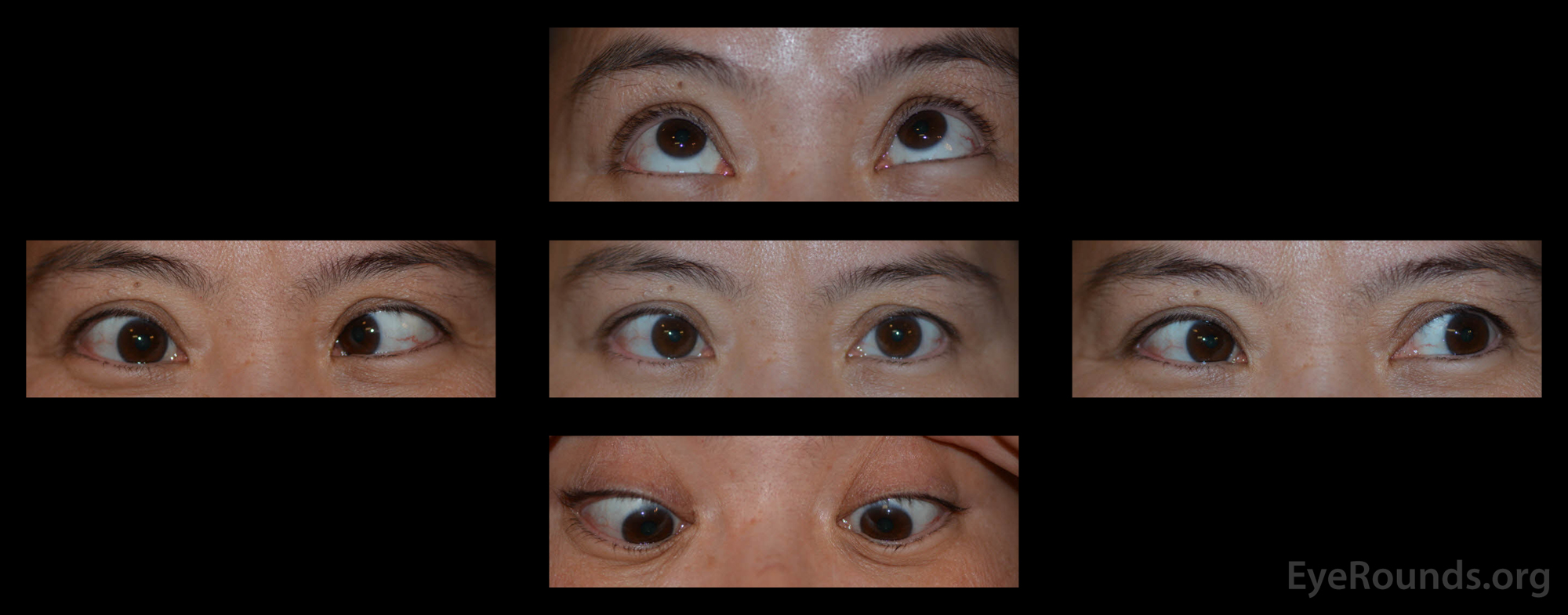
Figure 1. External photographs in primary position and several cardinal gazes. The patient had a large incomitant esotropia that increased on rightward gaze with an associated severe abduction deficit OD. There was a small adduction deficit OD. Supraduction, infraduction OD were full. There were no motility deficits OS.There was no proptosis, ptosis, lid retraction, or periorbital edema of either eye.
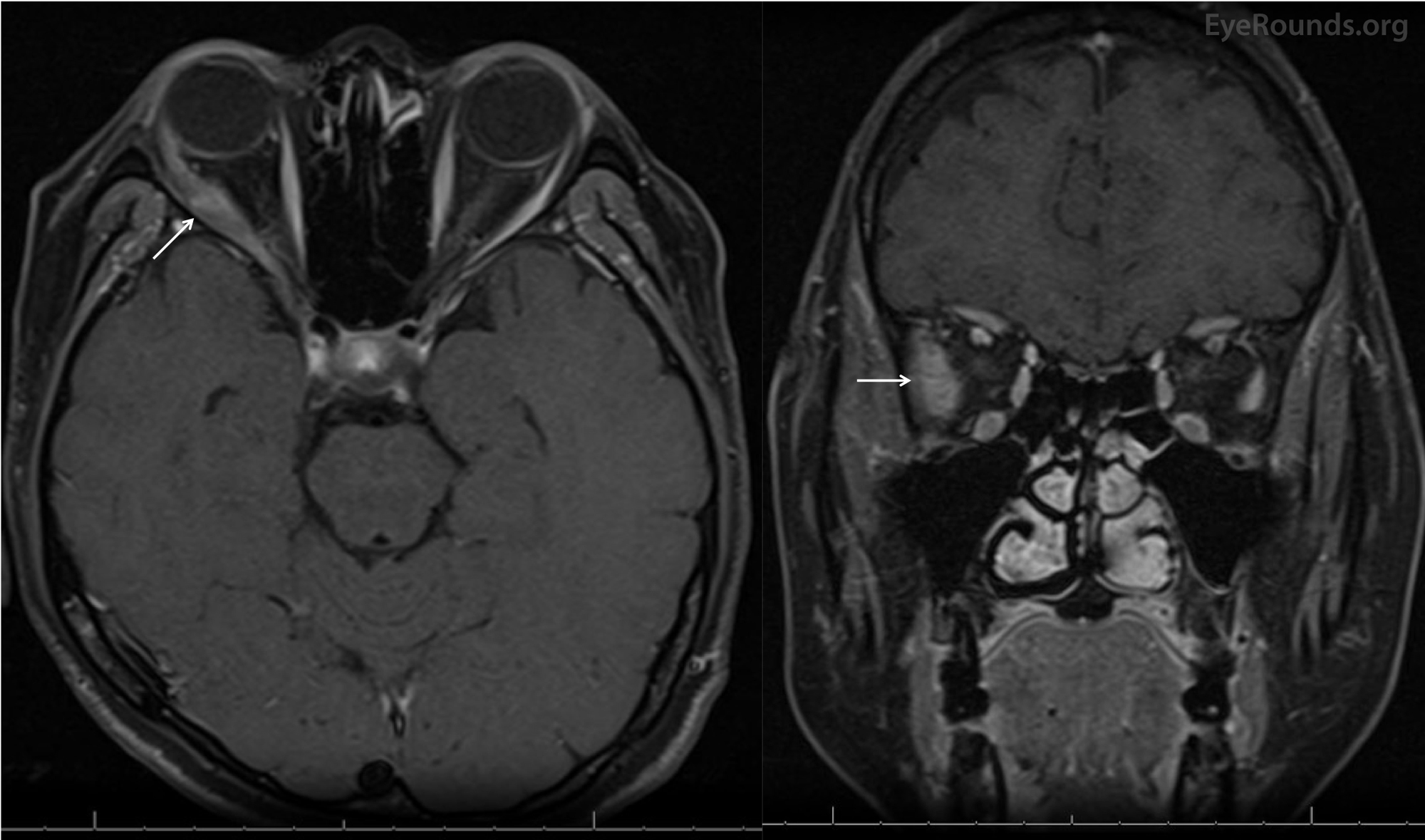
Figure 2. Magnetic resonance imaging (MRI) of the brain and orbits with contrast. Axial (left) and coronal (right) MR imaging showed significant non-uniform enlargement of the right lateral rectus muscle belly (white arrows) without enlargement of other extraocular muscle bellies or the lacrimal glands. There was no lesion along the course of the right sixth nerve. There was absence of sinus disease, retro-orbital mass, or enlargement of the lacrimal gland.
Differential Diagnosis of Isolated Extraocular Muscle Enlargement and Enhancement:
WORK-UP
Right lateral rectus biopsy was performed via a transconjunctival lateral orbitotomy
This patient had no significant medical history and an acute onset abduction deficit OD. This was originally thought to be due to microvascular ischemia due to a reportedly unremarkable MRI of the brain. However, the abduction deficit failed to recover. The large incomitant esotropia increased over time with worsening of her abduction deficit, especially on right gaze. On presentation to UIHC, the MRI was reviewed and showed significant, non-uniform enlargement of the right lateral rectus muscle belly (Figure 2). Lack of pain suggested that this was not an inflammatory process. The clinical history and imaging prompted the oculoplastics team to perform a laboratory work-up (see above) and to biopsy the lateral rectus. The histopathology was consistent with amyloidosis of the lateral rectus OD (Figure 3).
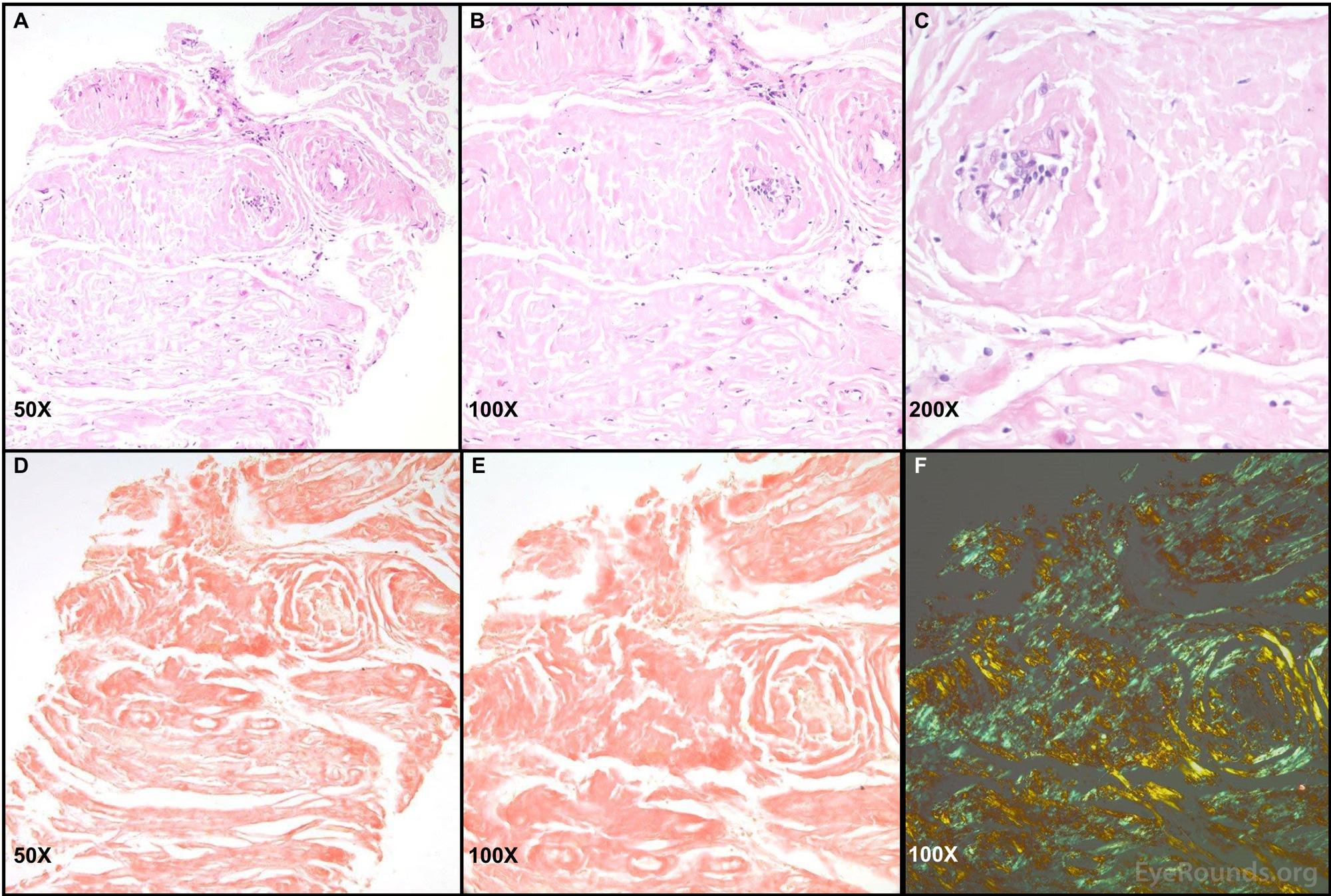
Figure 3. Right lateral rectus biopsy sample with Congo red stain and birefringence. Images A-C: Biopsy sections of the patient's lateral rectus demonstrated fragments of eosinophilic material that appeared largely collagenous. Areas of glassy pink material were present within the tissue. Only a few cells and degenerating muscle fibers were present. Images D and E: Congo red stain was positive throughout most of the tissue. Image F: Apple-green birefringence was identified when the Congo red stained tissue was visualized under polarized light. All tissue samples were processed, stained, and imaged by Dr. Nasreen Syed; these images have been used with her permission.
Hematology was consulted for evaluation to determine if amyloidosis was localized or systemic before proceeding with further therapy. Further work-up included positron emission tomography (PET) imaging, repeat MRI of the brain and orbits, bone marrow biopsy with serologies, echocardiogram, electrocardiogram, pulmonary function testing, an infectious disease panel for additional staining of the biopsy sample, mass spectroscopy of the rectus muscle, and genetic testing for familial amyloidosis. The bone marrow sample showed 3% polyclonal plasma cells. Cytogenetics studies and fluorescent in situ hybridization (FISH) analyses were normal.
The final diagnosis was localized AL amyloidosis of the right lateral rectus muscle. Follow-up visits with ophthalmology showed stable diplopia for two years. The radiation-oncology team considered radiation therapy but the patient was concerned about potential side effects of the treatment. Chemotherapy was deemed too risky given the lack of systemic symptoms. Therefore, it was decided that her condition would be managed with strabismus surgery. She underwent a 6 mm right medial rectus recession. Of note there was no restriction of the left lateral rectus on forced duction testing during the case. At the one-month follow-up appointment (Figure 4), she was doing well with no diplopia in primary gaze and an acceptable improvement in her head turn.

Post-operative strabismus testing after a right medial rectus recession. The patient had a small exophoria in primary gaze both in the distance and at near. She had a slight improvement of her abduction deficit OD.
Localized orbital amyloidosis
Amyloidosis is a heterogeneous group of diseases that occurs when the abnormal folding of proteins leads to a buildup of insoluble, extracellular amyloid fibrils, which are arranged in beta-pleated sheets. These fibrils can be deposited in various tissues throughout the body. The most common type of amyloidosis is amyloid light chain (AL) amyloidosis, which was formerly called primary amyloidosis; this occurs secondary to an underlying clonal proliferation, such as benign monoclonal gammopathies, multiple myeloma, or other B-cell disorders. The monoclonal immunoglobulin light chains deposit as AL amyloid, which damages tissues and causes the associated symptoms.
AL amyloidosis can be further divided into systemic and localized types. Systemic amyloidosis is more common and is a serious condition that can be fatal secondary to circulating amyloid deposition that often causes damage to many organ systems [1]. Localized amyloidosis is a much rarer form of the disease and is characterized by amyloid deposits limited to only the involved organ without systemic circulating monoclonal proteins or clonal plasmacytosis of the bone marrow. Common locations of the deposits include the gastrointestinal or genitourinary tracts, respiratory tract, orbits, skin, or cardiac tissue. This diagnosis carries an excellent prognosis for mortality, but the affected organs may sustain severe damage [2].
Rates of systemic amyloidosis have not been well characterized, but incidence of the most common type, AL amyloidosis, has been reported at 9 to 14 cases per million patients per year [3, 4]. The median age at diagnosis for systemic AL amyloidosis is 64 years and incidence increases with patient age [3, 4]. Men are most commonly affected, making up approximately 65% of new diagnoses [4], and we are unaware of any data showing race to be a factor in incidence rates for systemic disease.
There is a relationship between AL amyloidosis, which is due to monoclonal proliferations, and other plasma cell disorders. Monoclonal gammopathy of undetermined significance (MGUS), a clonal proliferation due to an unknown cause, precedes a diagnosis of AL amyloidosis in nearly all cases [5] but the frequency of conversion is less than 1% per year [6]. 10-15% of patients presenting with multiple myeloma are found to have coexistent AL amyloidosis [7]. Additionally, both localized and systemic amyloidosis can occur secondary to chronic auto-immune diseases [1, 2].
Localized amyloidosis is rare, and only 7-12% of patients newly diagnosed with any type of amyloidosis are found to have localized AL amyloidosis [2, 8]. It is presumed to be due to a local plasma cell clone dyscrasia [8].
While localized amyloidosis makes up only a small proportion of all amyloidosis cases, only 4% of localized cases involve the orbit, and of these, only 1.3% were found to have deposition in the extraocular muscles; when amyloidosis does involve the ocular tissues, it is most commonly found in the eyelid, followed by the orbit, conjunctiva, and lacrimal gland [9]. Involvement of the extraocular muscles, punctum, or levator palpebrae is extremely rare [9].
As in this case, extraocular motility deficits and diplopia resulting from extraocular muscle infiltration can be the presenting symptom of amyloidosis. Thus, amyloidosis should be considered in the differential for any patient with an atypical acquired strabismus. Patients may also report eye pain or discomfort, progressive bulging of the eye, blurry vision, and/or globe displacement, which may result from an orbital mass. If the lacrimal gland is involved, then the patient may experience dry eye and blurry vision. In most cases, localized orbital amyloidosis is unilateral [9].
On examination, extraocular motility deficits and binocular diplopia should be quantified. If there is an orbital mass, there may be decreased visual acuity, change in refractive error, and/or proptosis, which should be quantified with exophthalmometry. If there is compression of the optic nerve, there may be a relative afferent pupillary defect, which can be quantified using neutral density filters. Localized orbital amyloidosis may coexist with adnexal involvement. If the eyelid or conjunctiva are infiltrated, then patients may experience lid ptosis. There may also be a palpable periocular or conjunctival mass [10].
In the setting of localized orbital amyloidosis, a thorough review of systems and examination should be performed to rule out systemic involvement. Patients may present with generalized, non-specific symptoms of fatigue, weight loss, weakness, and signs and symptoms may vary widely depending on the organ system involved, as below.
Signs and symptoms of systemic amyloidosis, derived from Harrison's Principles of Internal Medicine, Chapter 108 on Amyloidosis [11]:
Systemic amyloidosis must be ruled out in the setting of localized orbital amyloidosis with a careful review of systems and examination to guide further work-up.
As amyloidosis is commonly reported in association with serious diseases involving clonal proliferation and is more commonly systemic rather than localized, any patient with evidence of localized amyloid accumulation should be evaluated for associated systemic disease. If there is evidence of circulating monoclonal proteins, there must be an evaluation for other sites of disease. This includes liver function tests, coagulation parameters, 24-hour urine protein electrophoresis, creatinine and albumin, glomerular filtration rate, troponin, NT-proBNP, echocardiogram, and electrocardiogram. Fat pad aspirate and bone marrow biopsy should be obtained (see below), and if these are negative, biopsies of affected organs should be obtained.
The bone marrow sample should be sent for the following:
As shown in Figure 3, the Congo red dye readily binds to tissue with amyloid deposition, and when viewed under polarized light, this stained tissue demonstrates apple-green birefringence. Hematoxylin and eosin staining shows abundance of eosinophilic material appearing largely collagenous.
Recent studies have shown promising treatments for systemic amyloidosis that include chemotherapy with high doses of melphalan, steroids and autologous stem cell transplantation [12]. Management for localized orbital amyloidosis has included observation alone, surgical debulking, and radiotherapy. Khaira, et al. described two patients with localized orbital amyloidosis and their response to surgical debulking followed by 30 Gy and 34 Gy fractionated external beam radiotherapy [13]. These researchers found that radiotherapy following debulking appears to halt the progression of the disease [13]. Copperman, et al. found that radiation therapy is effective for halting the progression of localized periocular AL amyloidosis at one year [14].
EPIDEMIOLOGY OR ETIOLOGYSystemic Amyloidosis
Localized Orbital Amyloidosis
|
SIGNSSystemic Amyloidosis [11, 12]
Localized Ocular Amyloidosis [9,10]
|
SYMPTOMSSystemic Amyloidosis
Localized Ocular Amyloidosis [9,10]
|
TREATMENT/MANAGEMENT
Systemic Amyloidosis
Localized Orbital Amyloidosis
|
Kennedy S, Scruggs BA, Fox AR, Larson SA, Shriver EM. Localized Orbital Amyloidosis Presenting as New-Onset Diplopia. EyeRounds.org. posted August, 13, 2018; Available from https://eyerounds.org/cases/278-orbital-amyloidosis.htm.

Ophthalmic Atlas Images by EyeRounds.org, The University of Iowa are licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License.

Address
University of IowaLegal
Related Links
