

Multiple Evanescent White Dot Syndrome
Authors: Mahsaw Motlagh, MD; Ryan J. Diel, MD; Elliott H. Sohn, MD
Posted January 5, 2022
CASE 1
INITIAL PRESENTATION
Chief Complaint
Blurry vision in the right eye
History of Present Illness
A 19-year-old woman presented with acute onset blurry vision in the right eye with associated changes in color vision and intermittent central photopsias. She could not identify any triggers or events related to the onset of her symptoms, which had started 10 days prior. She denied eye pain, pain with eye movements, or photophobia. Other than an upper respiratory infection about a month prior, she was healthy before the onset of her visual symptoms. Initial MRI of the orbits showed no evidence of optic neuritis or compressive lesion.
Past Ocular History
- Hyperopia with astigmatism of both eyes
- Single episode of anterior uveitis affecting the left eye six months prior
Past Medical History
- Asthma
- Seasonal allergies
- Iron deficiency anemia
- Generalized anxiety disorder
Medications
- Cetirizine
- Escitalopram
- Norgestimate-ethinyl estradiol
Allergies
- Sulfonamide antibiotics
Family History
- Nyctalopia in mother
Social History
- No history of tobacco or illicit drug use. Social alcohol use.
Review of Systems
- Negative unless otherwise noted in the history of present illness or above.
OCULAR EXAMINATION
Corrected visual Acuity
- Right eye (OD): 20/200, no improvement with pinhole
- Left eye (OS): 20/20
Ocular Motility/Alignment
- OD: Full extraocular motility
- OS: Full extraocular motility
Intraocular Pressure
- OD: 15 mm Hg by Tonopen
- OS: 14 mm Hg by Tonopen
Pupils
- OD: 7 mm in dark, 6 mm in light, 0.9 to 1.2 log unit relative afferent pupillary defect (RAPD)
- OS: 7 mm in dark, 6 mm in light
Confrontational visual fields
- OD: Partial superotemporal and inferotemporal restrictions
- OS: Full
External
- Both eyes (OU): Normal
Slit lamp exam
- Lids/lashes: Normal OU
- Conjunctiva/sclera: Clear and quiet OU
- Cornea: Clear OU
- Anterior Chamber: Deep and quiet OU
- Iris: Normal architecture OU
- Lens: Trace nuclear and cortical cataract OU
Dilated fundus examination (DFE)
- Vitreous: Normal, no vitritis OU
- Disc:
- OD: Normal
- OS: Normal
- Cup-to-disc: Small, congested discs OU
- Macula:
- OD: Orange granular appearance of the fovea, multiple faint circular white lesions throughout the posterior pole most concentrated in the temporal macula
- OS: Normal
- Vessels: Normal OU
- Periphery: Normal OU
Additional testing
Goldmann visual field (GVF) testing
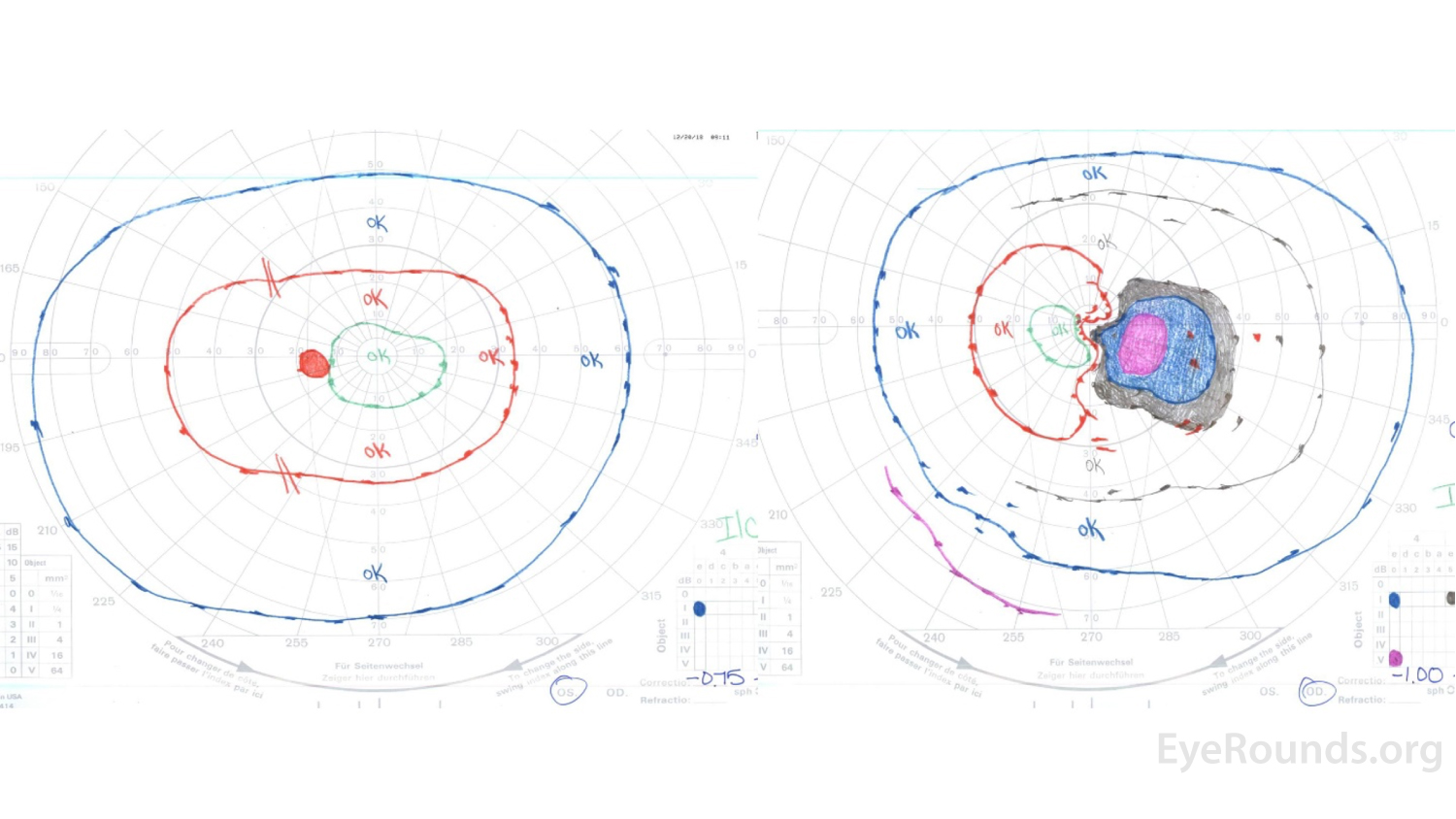
Figure 1: GVF of the right eye (right panel) revealed a markedly enlarged blind spot with cecocentral scotoma at the time of presentation. The left eye (left panel) was normal.
Color fundus photography
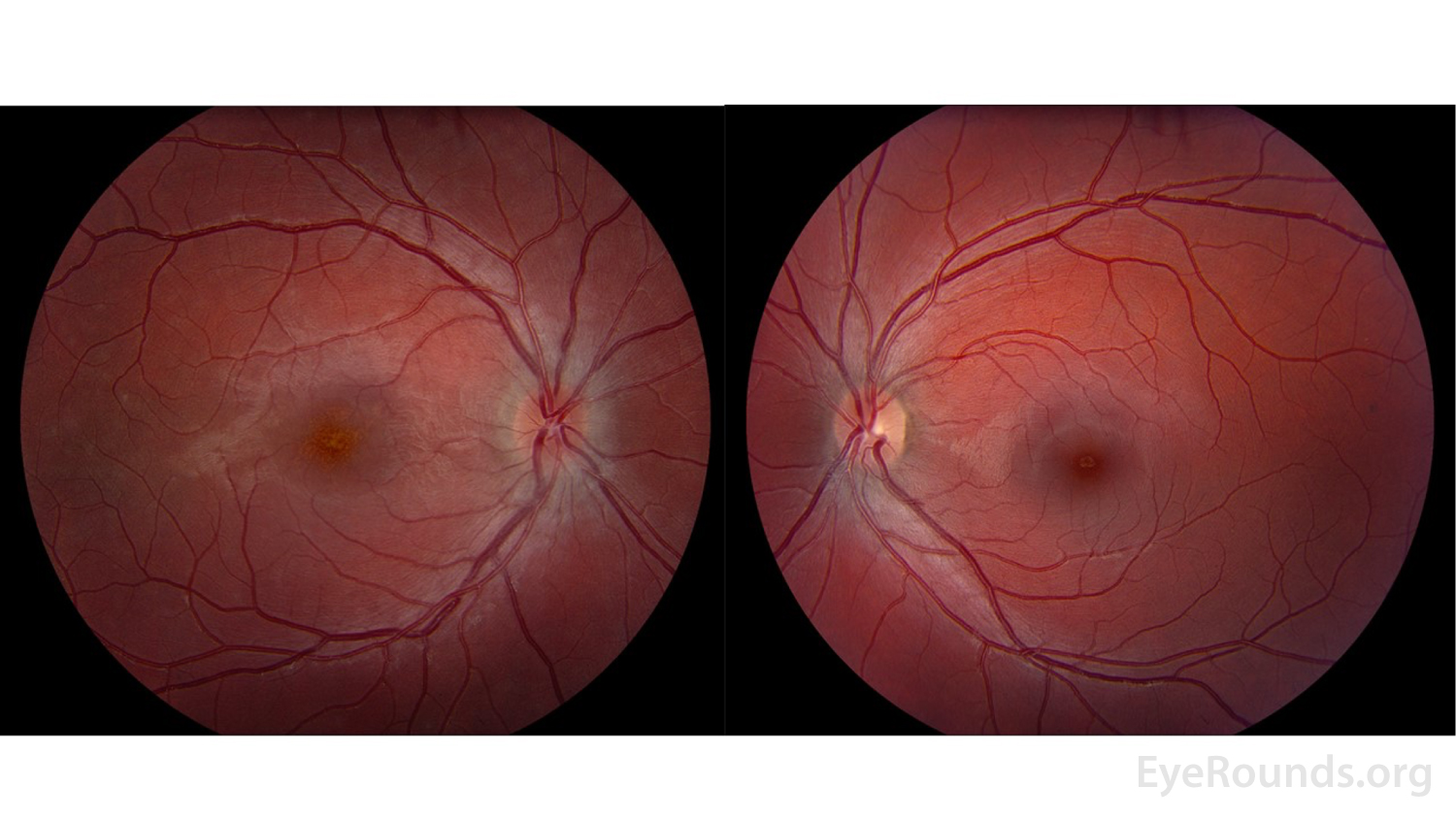
Figure 2: Color fundus photos of the right eye (left panel) demonstrated a small, structurally congested optic disc with mild optic disc edema in addition to an orange granular appearance of the macula. There were also multiple faint circular lesions throughout posterior pole, most prominent in the temporal macula. The left eye (right panel) similarly demonstrated a small, structurally congested optic disc but was otherwise normal.
Optical coherence tomography (OCT) macula
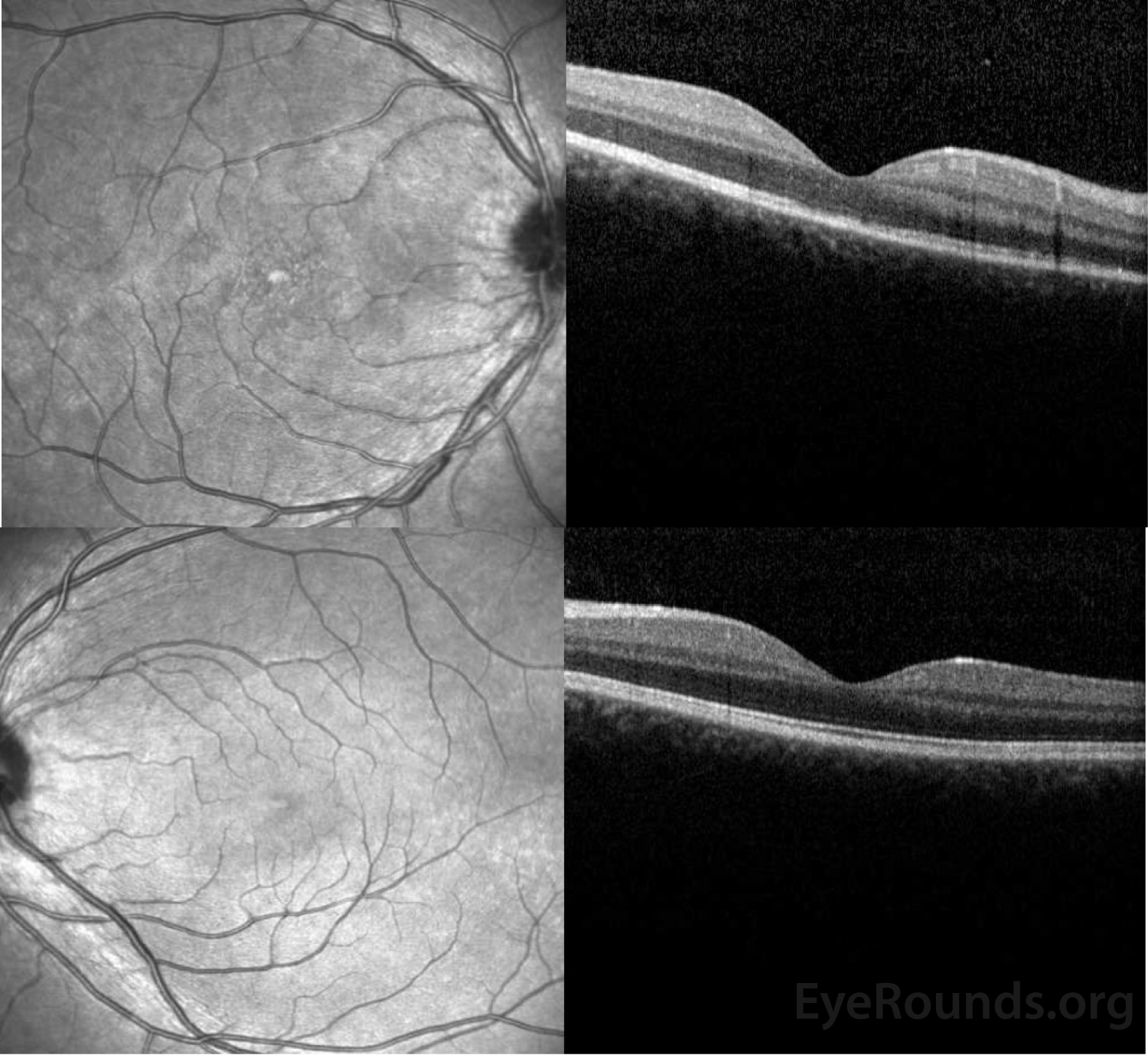
Figure 3: OCT of the right eye (top panel) shows a subtle stippled inner segment/outer segment (IS/OS) junction and normal foveal contour without edema. The left eye (bottom panel) shows normal foveal contour and normal retinal architecture.
Fundus autofluorescence (FAF)
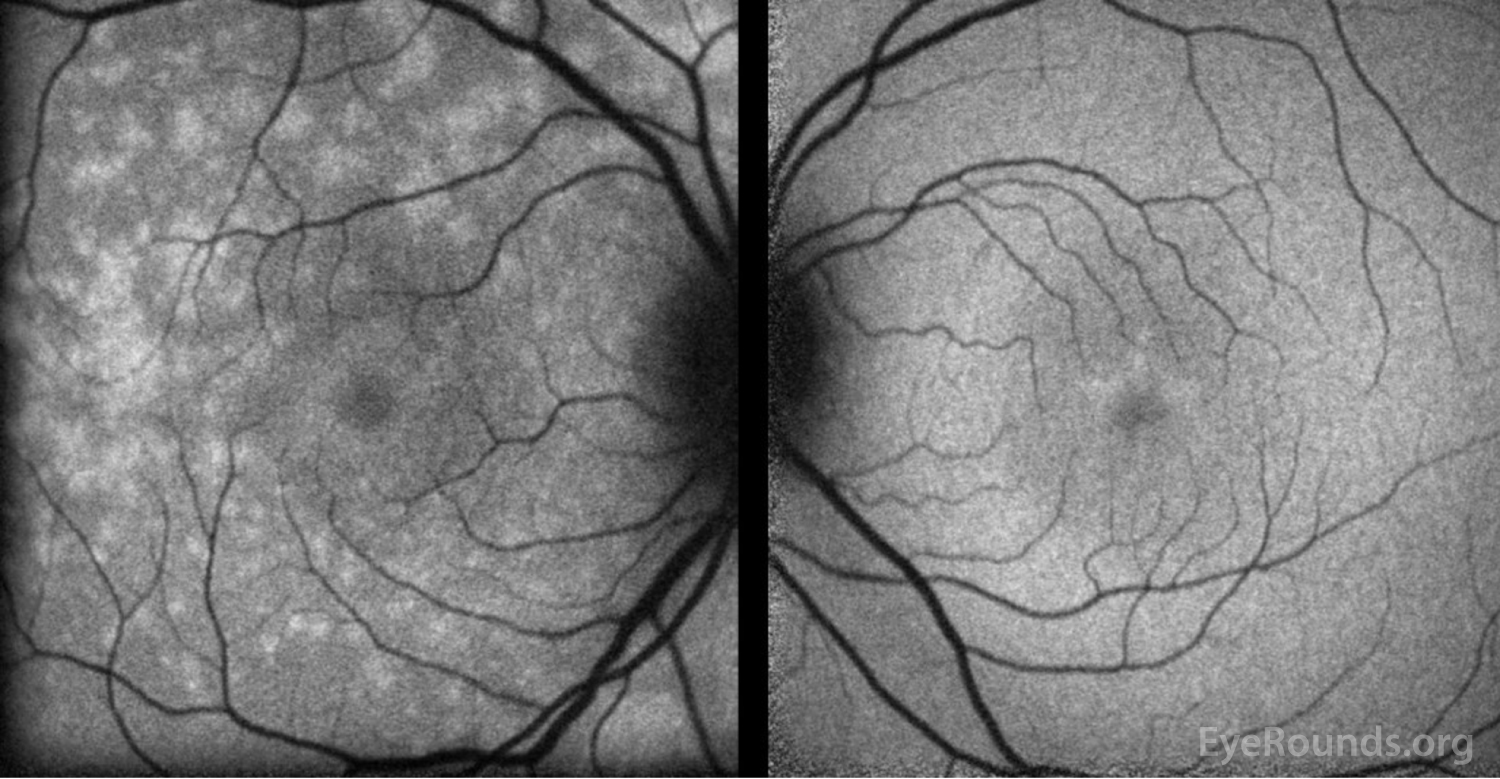
Figure 4: Fundus autofluorescence of the right eye (left panel)shows multiple foci of hyperautofluorescence throughout the posterior pole concentrated most in the temporal macula while the left eye (right panel) is relatively normal.
Fluorescein angiography (FA)
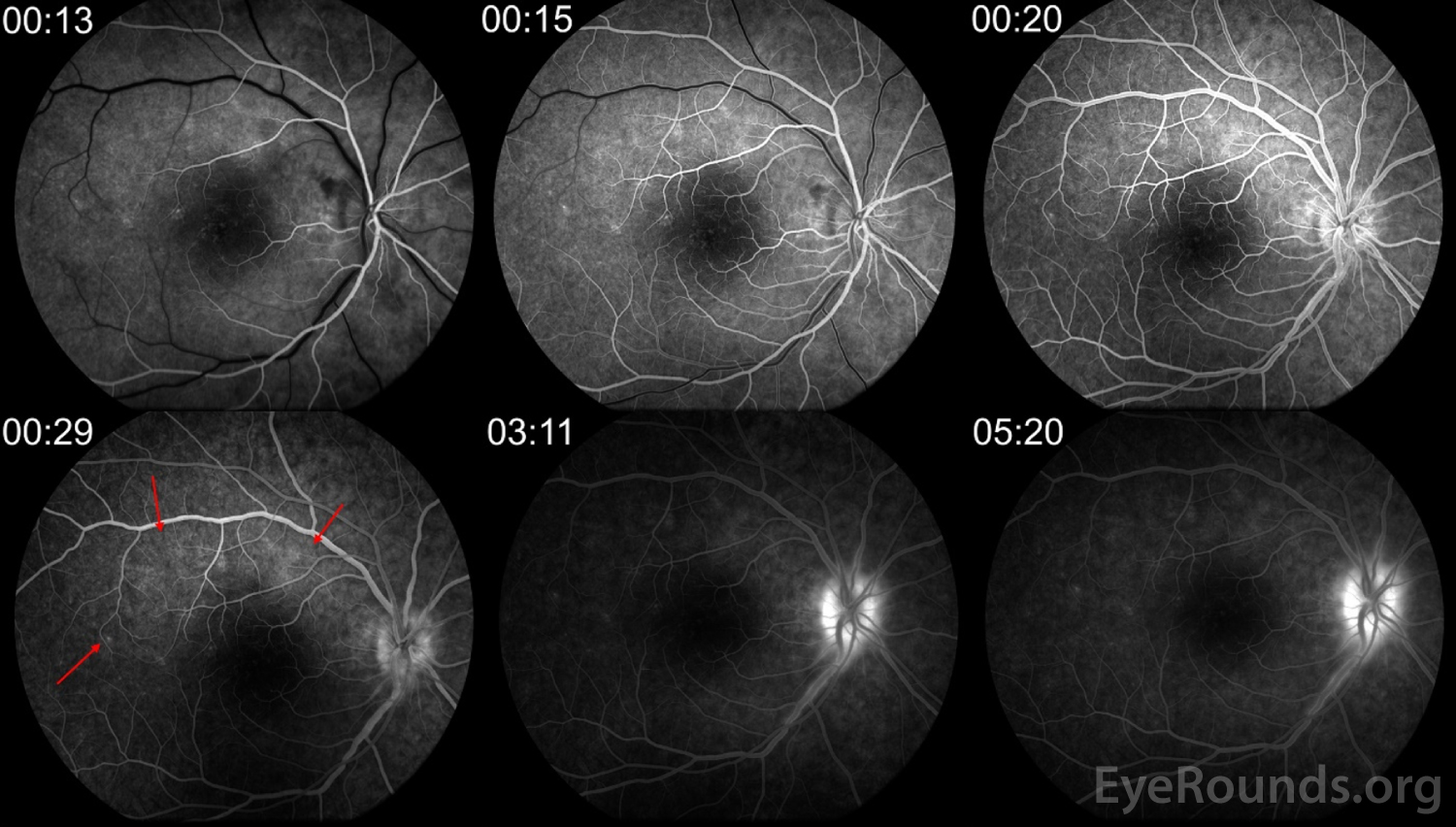
Figure 5: FA of the right eye demonstrates multiple small foci of early and late hyperfluorescence in a wreath-like pattern (red arrows) corresponding to the lesions seen on FAF.
Multifocal Electroretinogram (mfERG)
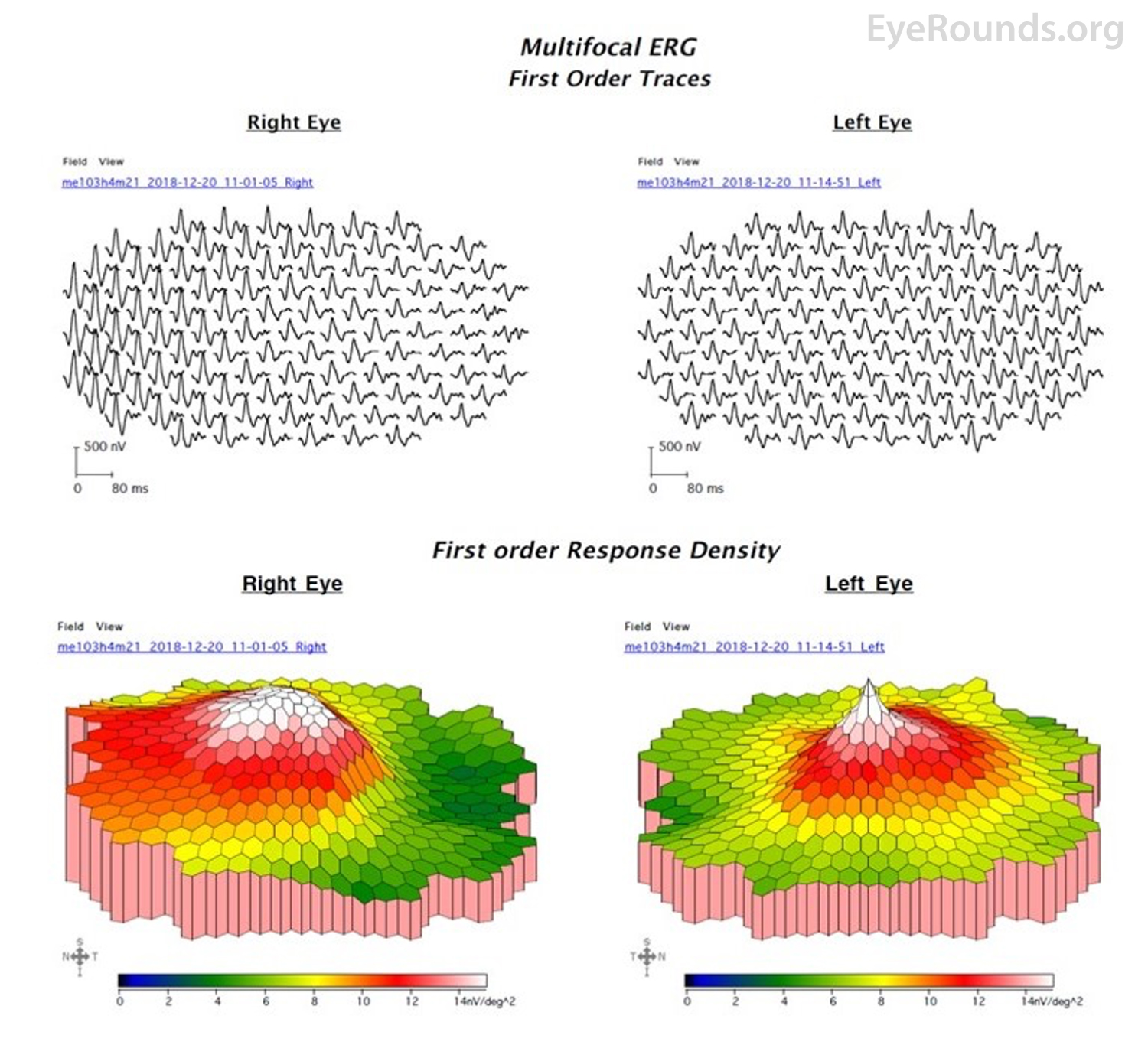
Figure 6: mfERG of the right eye at presentation demonstrated increased amplitudes in the temporal macula, while the left eye was relatively normal.
Differential Diagnosis
- Multiple evanescent white dot syndrome
- Acute idiopathic blind spot enlargement syndrome
- Acute zonal occult outer retinopathy
- Punctate inner choroidopathy
- Optic neuritis
Diagnosis
- Multiple evanescent white dot syndrome with optic nerve involvement
CLINICAL COURSE
After ruling out optic neuritis via initial MRI and being given a diagnosis of MEWDS and optic nerve involvement with APD, the patient was referred to rheumatology for a complete laboratory work-up for autoimmune etiologies. In the interim, she was started on oral steroid therapy. Autoimmune work-up revealed an elevated antinuclear antibody of 1:160 with a nuclear fine speckled pattern. All additional work-up including anti-SSA antibody, anti-SSB antibody, beta-2 microglobulin, rheumatoid factor, C-reactive protein, erythrocyte sedimentation rate, and angiotensin-converting enzyme were negative or normal. Infectious work-up for Toxoplasmosis, tuberculosis, syphilis, Lyme disease, Bartonella, HIV, and Chlamydia were all negative. Unfortunately, at her two-week follow-up the patient's vision continued to worsen from 20/200 at initial presentation to 20/250. She was therefore transitioned to a steroid-sparing regimen of mycophenolate mofetil (CellCept) under the care of rheumatology. Her vision later recovered over the course of six months and she was gradually tapered off therapy without recurrence. Since then, she continues to have resolution of the cecocentral scotoma and prior imaging findings.
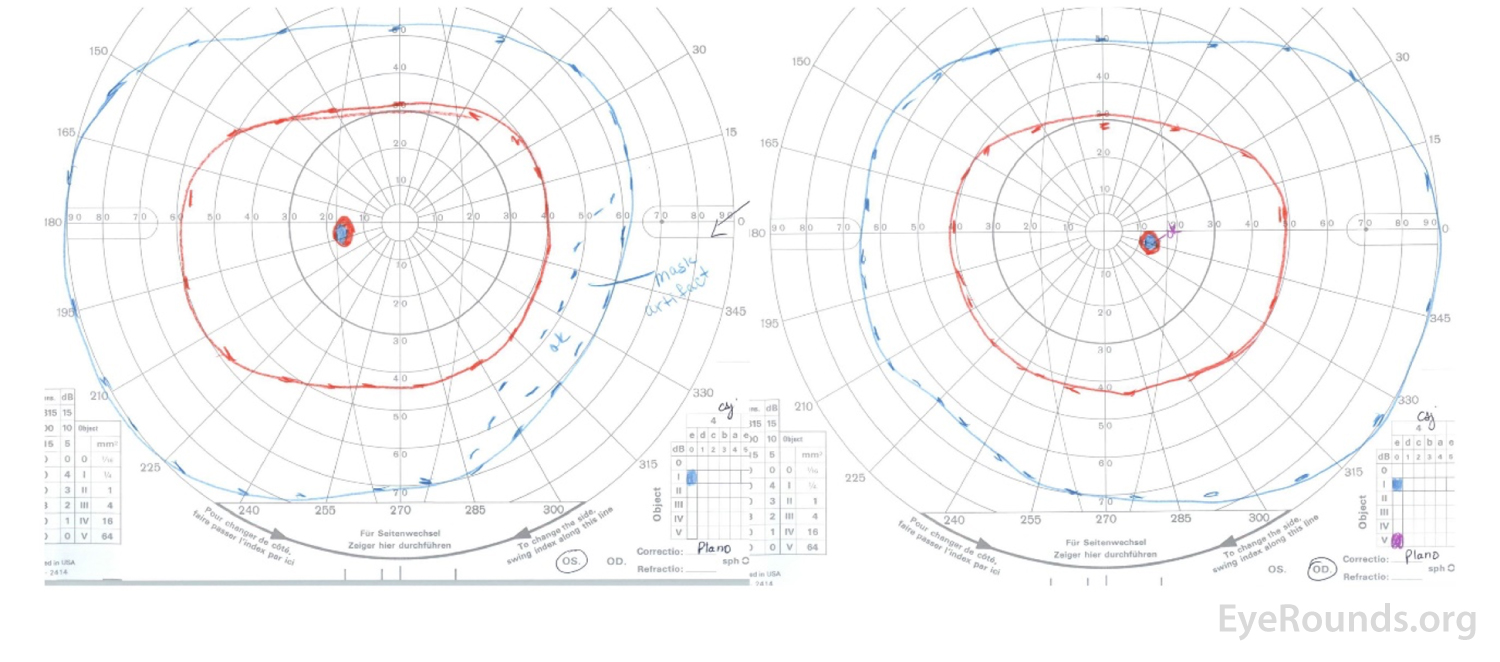
Figure 7: GVF after treatment demonstrating resolution of the visual field deficits in the right eye.
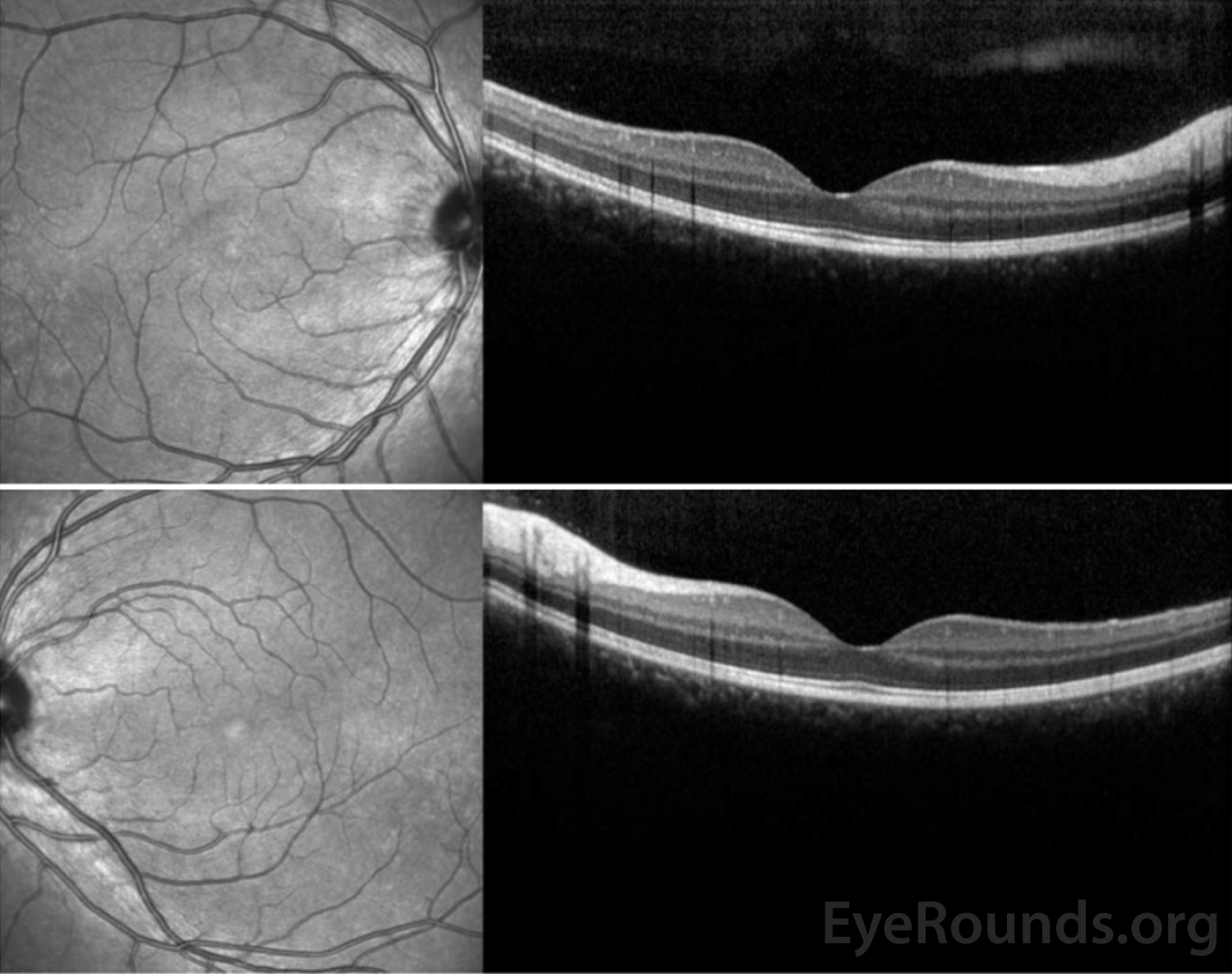
Figure 8: OCT of the right eye (top panel) demonstrated resolution of the previous IS/OS stippling and the left eye (bottom panel) remained normal.
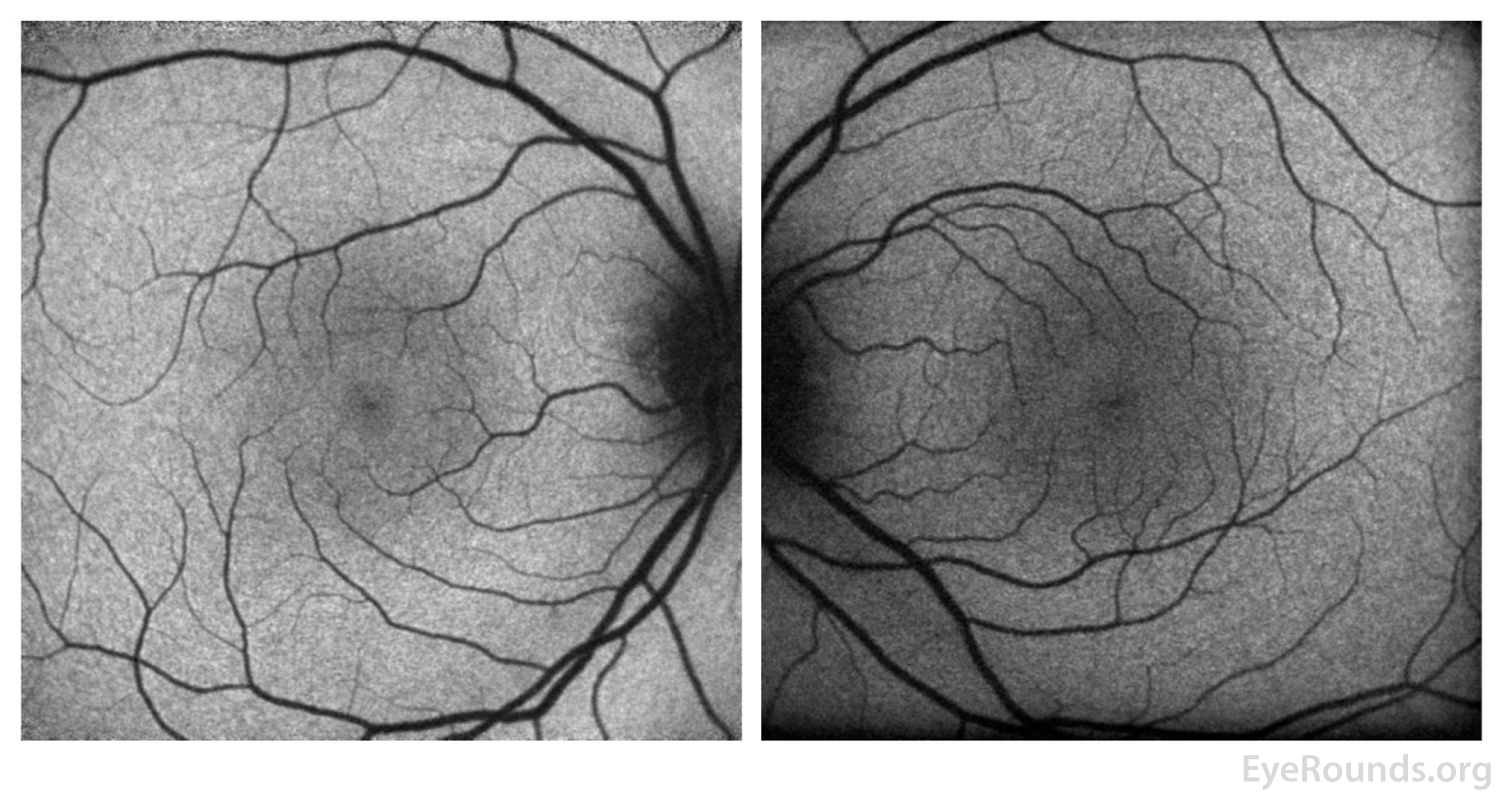
Figure 9: FAF of the right eye (left panel)shows resolution of the previous foci of hyperautofluorescence and the left eye (right panel) remains normal.
CASE 2
INITIAL PRESENTATION
Chief Complaint
Gray spot in the center of his vision in the right eye
History of Present Illness
A 44-year-old Japanese man presented for evaluation of a "gray spot" involving his central vision in the right eye. His symptoms started one week prior without an inciting trigger or event. The patient recalls a similar episode 8 years prior and states that he was diagnosed with a "white dot syndrome" at another facility. At that time, it took 8 weeks for his symptoms to resolve without treatment and he had been asymptomatic until now. He denied photopsias, metamorphopsia, or dyschromatopsia. He denied any recent illness or vaccinations.
Past Ocular History
- Unspecified white dot syndrome affecting the right eye
- High myopia (>10D) with astigmatism of both eyes
Past Medical History
- None
Medications
- None
Allergies
- No known allergies
Family History
- Non-contributory to current presentation
Social History
- Former smoker
Review of Systems
- Negative unless otherwise noted in the history of present illness or above
OCULAR EXAMINATION
Corrected visual Acuity
- Right eye (OD): 20/600, pinholes to 20/400
- Left eye (OS): 20/20
Ocular Motility/Alignment
- OD: Full extraocular motility
- OS: Full extraocular motility
Intraocular Pressure
- OD: 15 mm Hg by Tonopen
- OS: 13 mm Hg by Tonopen
Pupils
- OD: 3 mm in dark, 2 mm in light, no afferent pupillary defect (RAPD)
- OS: 3 mm in dark, 2 mm in light, no RAPD
Confrontational visual fields
- OD: Central scotoma
- OS: Full
External
- OU: Normal
Slit lamp exam
- Lids/lashes: Normal OU
- Conjunctiva/sclera: Clear and quiet OU
- Cornea: Clear OU
- Anterior Chamber: Deep and quiet OU
- Iris: Normal architecture OU
- Lens: Trace nuclear and cortical cataract OU
Dilated fundus examination (DFE)
- Vitreous:
- OD: Rare cell
- OS: Normal
- Disc: Tilted with peripapillary atrophy OU
- Cup-to-disc: Small, congested discs OU
- Macula:
- OD: Fine orange granularity noted in central macula
- OS: Normal
- Vessels: Normal OU
- Periphery: Normal OU
Additional testing
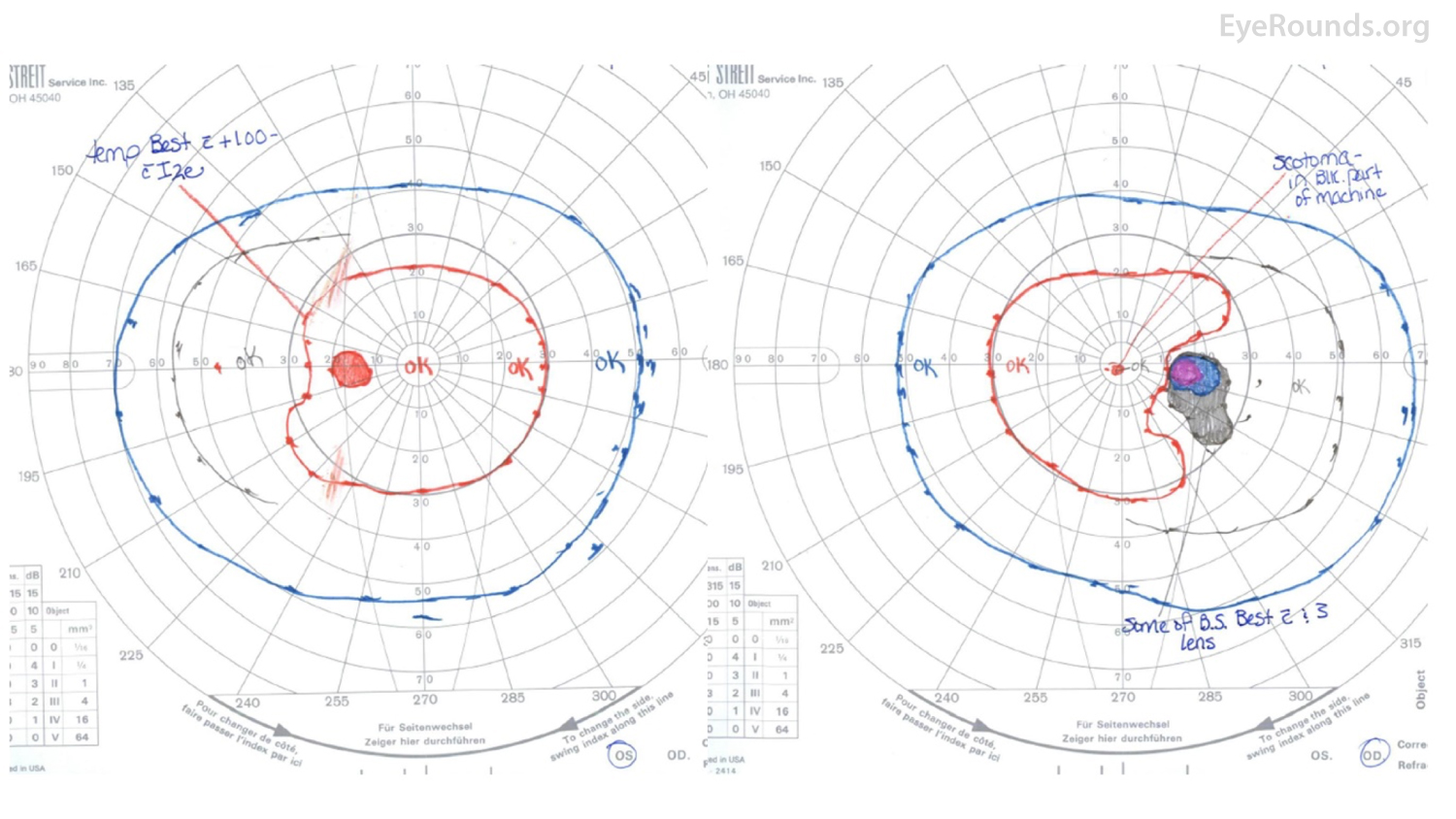
Figure 1: GVF of the right eye (right panel) demonstrates a markedly enlarged blind spot, while the left eye (left panel) is full.
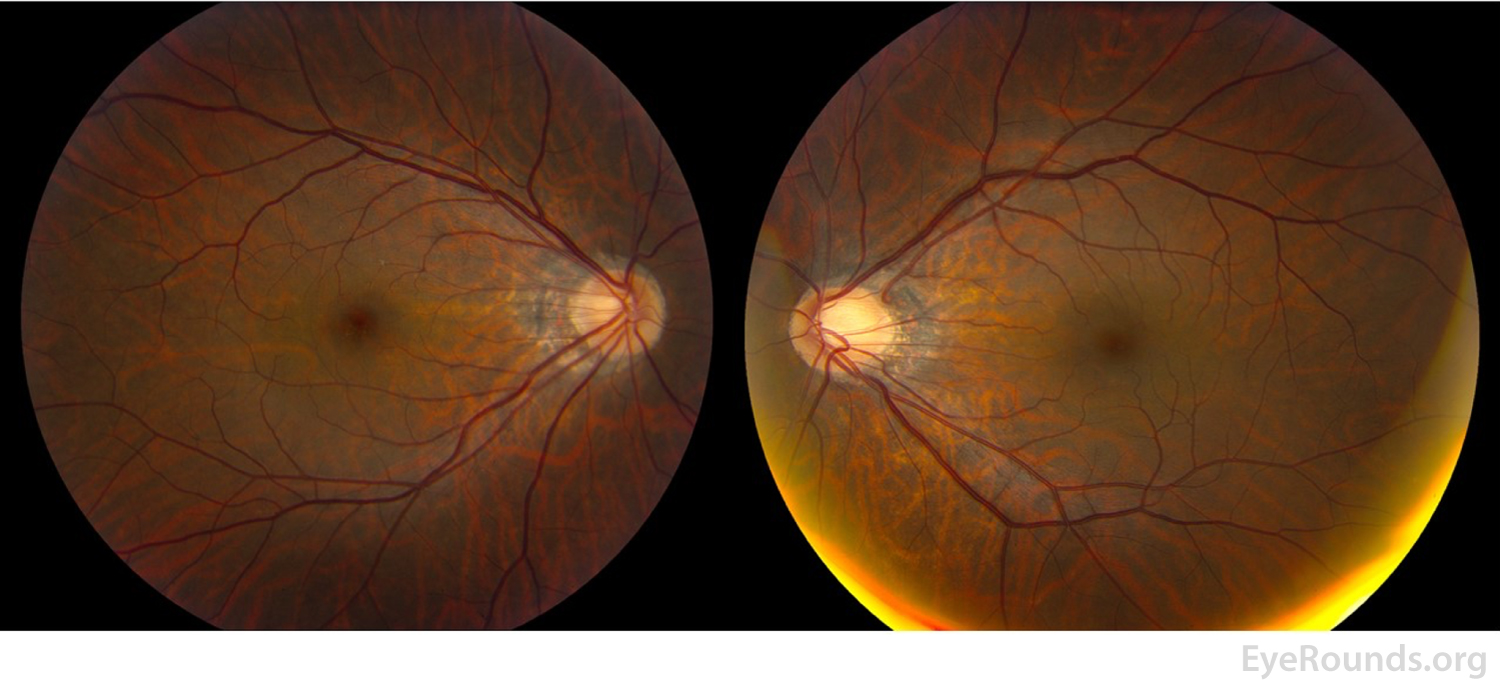
Figure 2: Color fundus photography of the right eye (left panel) shows subtle foveal granularity and the left eye is normal (right panel).
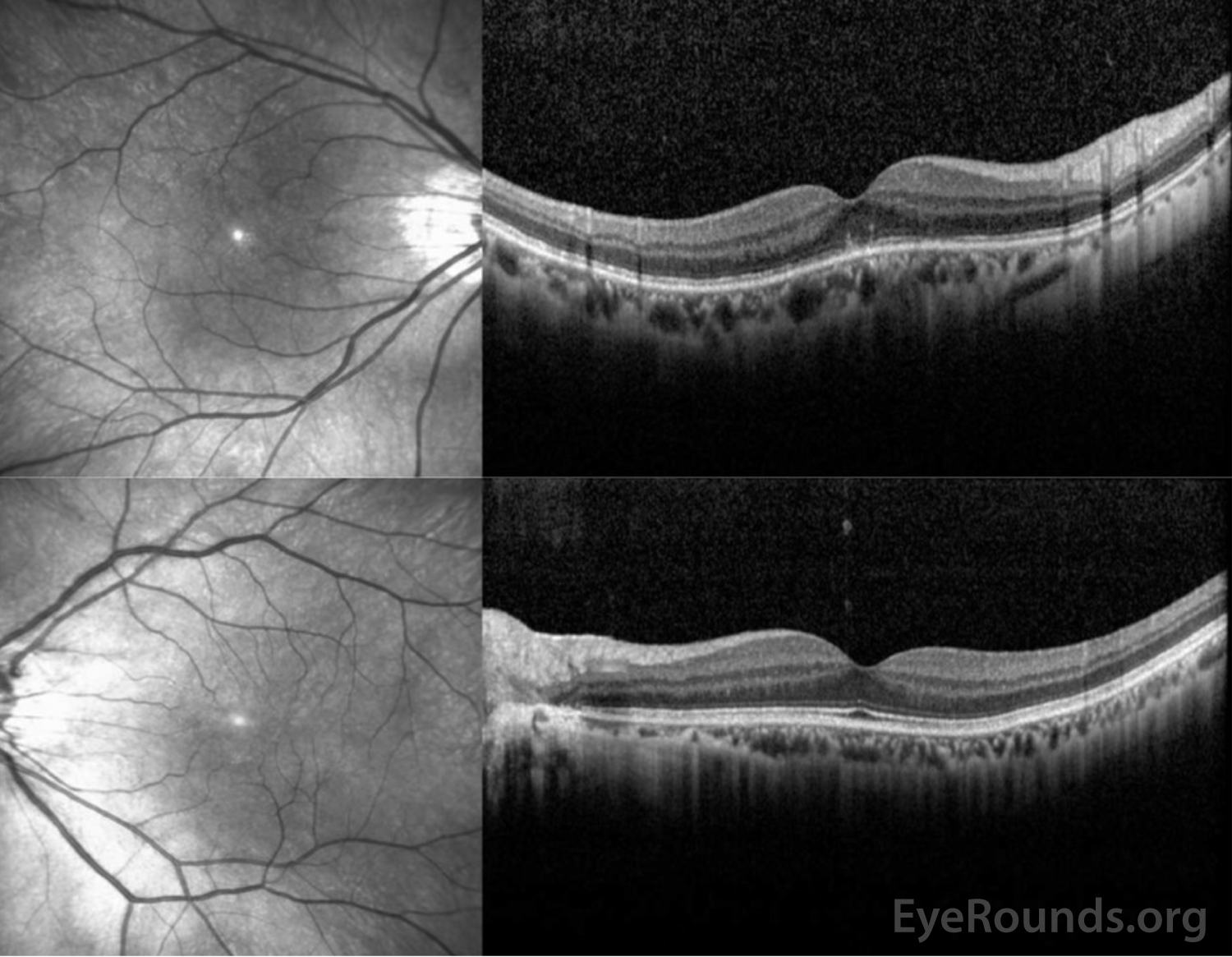
Figure 3: OCT of the right eye (top panel) shows a normal retinal contour but with focal hyperreflective abnormalities in the outer retina disrupting the ellipsoid zone centrally. The left eye (bottom panel) demonstrates normal retinal contour and striations.
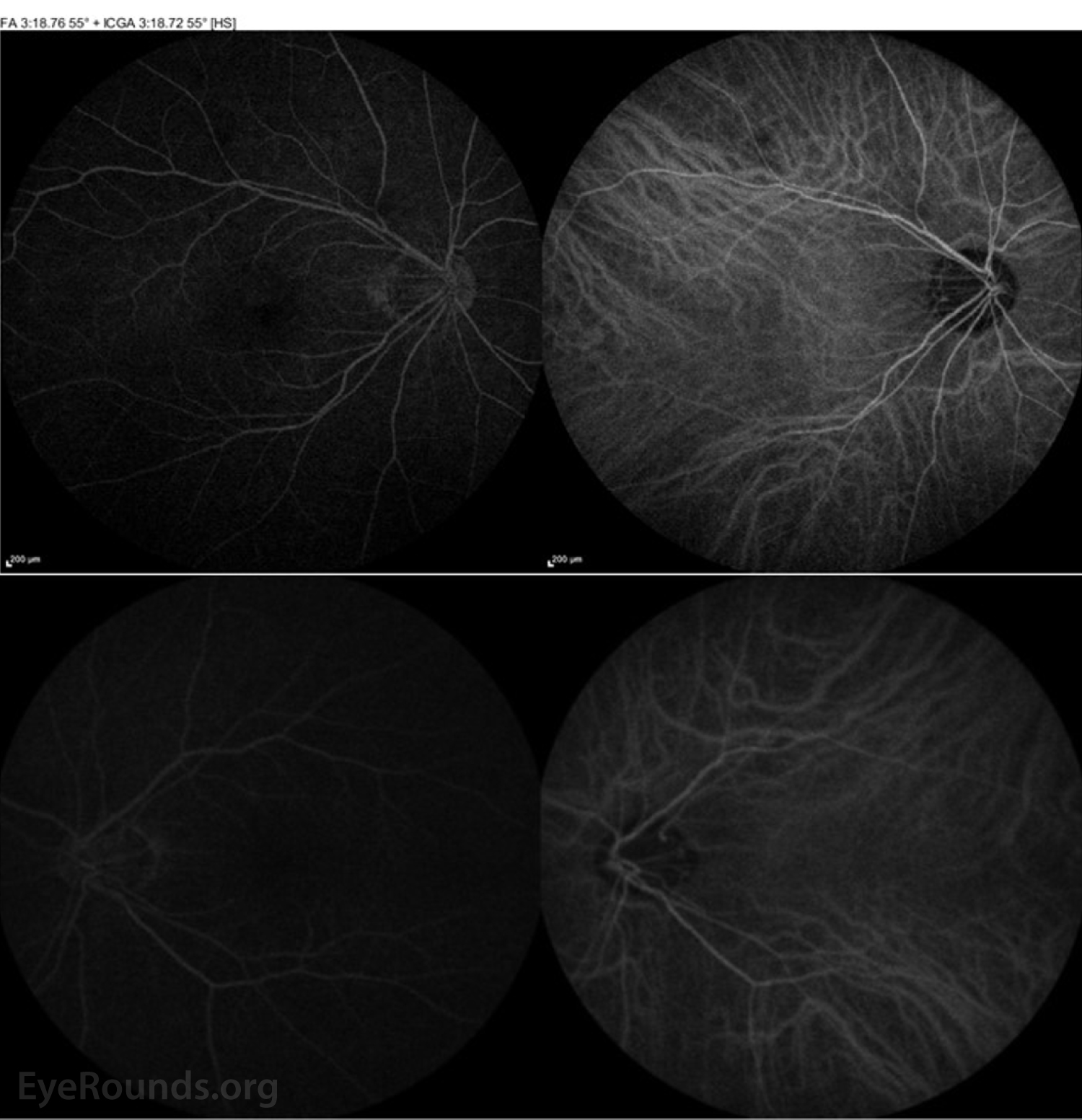
Figure 4: ICGA of both the right eye (top panel) and left eye (bottom panel) were normal.
Differential Diagnosis
- Multiple evanescent white dot syndrome
- Multifocal choroiditis
- Acute posterior multifocal placoid pigment epitheliopathy
- Punctate inner choroidopathy
- Birdshot chorioretinopathy
- Acute macular neuroretinopathy
- Acute zonal occult outer retinopathy
Diagnosis
Multiple evanescent white dot syndrome
CLINICAL COURSE
In the context of the patient's history and current clinical presentation, a diagnosis of recurrent MEWDS was made. The patient was provided reassurance and no treatment was initiated. Prior to his six-week follow up, the patient's symptoms had entirely resolved, so he cancelled his appointment. He represented in optometry clinic 6 months later for a contact lens evaluation and his vision had returned to 20/20 in both eyes. There was resolution of funduscopic findings and therefore further imaging was deferred.
DISCUSSION
Etiology/Epidemiology
The description of multiple evanescent white dot syndrome (MEWDS) was first published in 1984 as an idiopathic chorioretinal disorder.[1, 2] It is more common in young to middle-aged myopic females with a reported 4:1 female to male predilection.[3] However, patients with MEWDS have been reported even up to their seventh decade of life.[4] MEWDS generally follows an acute presentation and manifests almost always in a unilateral fashion.[1] It is unclear what precipitates the onset of disease, but it has been associated with a viral prodrome and recent vaccination.[1] There is no racial or hereditary predilection, though there is the potential for an underlying autoimmune disease. HLA-B51 has been reported at a 3.7-fold increased frequency in patients with MEWDS.[5]
Pathophysiology
The cause of MEWDS is unknown. Advances in retinal imaging with optical coherence tomography angiography (OCTA) and en face optical coherent tomography (OCT) point towards a primary disruption of the photoreceptors rather than that of the choriocapillaris.[6-8] OCT has clearly demonstrated that the lesions seen in MEWDS are localized to the ellipsoid zone and outer nuclear layer.[6, 7] Furthermore, studies with OCTA clearly demonstrate preservation of flow within the choriocapillaris [9, 10], reiterating the primary disruption in MEWDS occurs at and around the ellipsoid zone.
Fundus autofluorescence imaging (FAF) can demonstrate areas of hyperautofluorescence (hyperAF) in the early stage of disease that correlate to defects seen on indocyanine green angiography (ICGA) and fluorescein angiography (FA). Similar to resolution of lesions often seen with OCT, hyperAF spots typically resolve within several months.[11] What remains to be reconciled is the presence of hypofluorescent lesions on ICGA, which typically translates to choroidal non-perfusion. It is speculated that ICGA findings in MEWDS are actually better explained by secondary RPE disruption, altered reflectivity of the RPE, and possibly impaired uptake of the ICG pigment.[6, 7] Even with the exact underlying mechanism unclear, the acute disruption of photoreceptor outer segments is uniformly transient.[1] There is likely an interplay between the immune system and environmental triggers that precipitate an inflammatory response resulting in MEWDS.
In summary, the evolution in the understanding of the pathophysiology MEWDS points toward subtle disruption of the outer retina and photoreceptors without primary RPE disruption or choroidal nonperfusion. This shift has been the direct result of advancing retinal imaging techniques with perhaps the most important contribution coming from en face OCT, which specifically demonstrates diffuse and focal disruptions of the inner and outer segment junction.[6]
In regard to environmental triggers, MEWDS has been well associated with a viral prodrome[1] and after administration of vaccines including but not limited to influenza[12], rabies[13], yellow fever[14], meningococcal[15], human papilloma virus[15], and hepatitis A/B.[14]
Signs/Symptoms
Despite an unclear understanding of the underlying cause of MEWDS, the clinical features are well described. Recently, the Standardization of Uveitis Nomenclature (SUN) group has proposed classification criteria for MEWDS that can be found in Table 1.[16] As a result of the transient disruption of the photoreceptors, presenting clinical signs are frequently central vision loss, enlarged blind spot, and photopsia. Patients may also endorse grey haze, metamorphopsia, or dyschromatopsia.
On clinical exam, the hallmark features of MEWDS are transient white dots in the posterior pole with possible extension into the mid-periphery. These discrete white lesions are at the level of the outer retina and are about 100-200 microns in size.[1] Fundus examination may also reveal macular pigmented stippling or yellowish foveal granularity, and may be the only presenting sign.[17] The anterior exam is generally unremarkable, but there may be subtle signs of ongoing inflammation. The remainder of the posterior exam may also reveal disc edema, peripapillary inflammation, or posterior vitritis.[1] Rarely, patients with MEWDS develop choroidal neovascularization after the acute lesions heal.[18] At any point in the disease course chorioretinal scars or RPE mottling may also be present but are rarely visually significant.
Clinical Criteria |
Multifocal chorioretinal gray-white spots with foveal granularity |
|
AND |
Characteristic FA or OCT findings: (a) "Wreath-like" hyperfluorescent lesions on FA OR (b) hyperreflective lesions on OCT extending from the RPE into and/or through the ellipsoid zone into the ONL of the retina. |
|
AND |
|
Absent to mild anterior chamber and vitreous inflammation |
Table 1. Classification criteria for MEWDS adopted from the SUN working group.[16] (FA = fluorescein angiogram; OCT = optical coherence tomography; ONL = outer nuclear layer; RPE = retinal pigment epithelium)
Testing/Imaging
Multimodal imaging has become invaluable in the evaluation and management of MEWDS. Fluorescein angiography demonstrates punctate hyperfluorescence in the early phase and late staining in a typical "wreath-like" pattern around the fovea.[11] Indocyanine green angiography demonstrates hypofluorescent dots in the late phase that are frequently more numerous than those appreciated with FA or on clinical examination.[11] Fundus autofluorescence demonstrates hyperAF areas that correspond to the hyperfluorescent spots on late phase FA and hypofluorescent spots on ICGA.[8] FAF has been suggested to be the most sensitive of all imaging modalities as it can reveal lesions in absence of any white dots on clinical exam.[19] Optical coherence tomography demonstrates focal areas of photoreceptor disruption or an irregular ellipsoid zone.[8] Additionally, hyperreflective material or spots may be visualized in the outer nuclear layer on OCT.[20]
In MEWDS, it has been proposed that lesions on multimodal imaging have been defined as two types: larger "spots" (approximately 200 microns in diameter) and smaller "dots" (less than 100 microns in diameter).[6] En face visualization of OCT is a reconstruction algorithm of standard OCT images that produces transverse images of retinal and choroidal layers (also called coronal or C-scan). This allows clinicians to view specific retinal layers from a "bird's eye view"[6], which has been exceedingly useful in characterizing these two types of lesions. With en face OCT the larger "spots" were hyporeflective and localized to the ellipsoid zone, while the smaller "dots" were hyperreflective and localized to the outer nuclear layer.[6]
When available, full field electroretinography (ffERG) or multifocal electroretinography (mfERG) demonstrate dysfunctional photoreceptors characterized by transiently reduced a-wave amplitude.[2] In the early course of disease, patients may often demonstrate supernormal first-order amplitudes in mfERG, which may correspond to the inflammatory dynamics of MEWDS.[21] Beyond two weeks in the disease course, amplitudes may be normal, low, or even nonrecordable. Importantly, clinical recovery corresponds with significant recovery of recorded amplitude response on ERG. This is a key difference from patients with multifocal choroiditis and acute zonal occult outer retinopathy, who typically do not demonstrate recovery of photoreceptor function on mfERG.
Lastly, Humphrey visual field testing or Goldmann visual field may reveal a markedly enlarged blind spot with possible central scotoma at the time of initial presentation the size of which depends on the area of focal photoreceptor disruption.
Even though MEWDS is usually a unilateral disease, it is important to conduct multimodal imaging tests on both eyes as bilateral disease can be silent.[22] Beyond multimodal imaging, in patients with a suggestive clinical history there should be consideration of and subsequent workup for an underlying autoimmune disease or simultaneous multifocal choroiditis.
Treatment/Management/Guidelines
The natural disease course of MEWDS is favorable, and the majority of patients recover fully.[8] While treatment is mainly expectant, there are several reports of expediting visual recovery with use of corticosteroid therapy.[23] Without any kind of treatment, recovery tends to occur over 6 to 10 weeks.[8] Recovery is objectively demonstrated with return of normal architecture and function on imaging modalities, which corresponds with clinical improvement of patients. Atypical cases such as those with prolonged course and/or other signs of ocular involvement such as case 1 presented above may benefit from anti-inflammatory treatment, though this is not typically needed for patients with MEWDS.
If patients with MEWDS develop the rare complication of choroidal neovascularization, the treatment of choice is intravitreal anti-vascular endothelial growth factor agents. Even after resolution of the acute phase, RPE mottling or pigmentary changes may persist. While most patients have a benign course, visual acuity can be impaired long-term. Younger age and worse presenting visual acuity have been associated with a worse visual outcome.[24] While recurrences of MEWDS are rare, they can occur as seen in case 2 above. Treatment for recurrences is also expectant with a similar timeline of resolution.
EPIDEMIOLOGY OR ETIOLOGY
|
SIGNS
|
SYMPTOMS
|
TREATMENT/MANAGEMENT
|
REFERENCES
- Jampol, L.M., et al., Multiple evanescent white dot syndrome. I. Clinical findings. Arch Ophthalmol, 1984. 102(5): p. 671-4.
- Sieving, P.A., et al., Multiple evanescent white dot syndrome. II. Electrophysiology of the photoreceptors during retinal pigment epithelial disease. Arch Ophthalmol, 1984. 102(5): p. 675-9.
- Gross, N.E., et al., Multiple evanescent white dot syndrome. Arch Ophthalmol, 2006. 124(4): p. 493-500.
- Lim, J.I., G.T. Kokame, and J.P. Douglas, Multiple evanescent white dot syndrome in older patients. Am J Ophthalmol, 1999. 127(6): p. 725-8.
- Borruat, F.X., et al., HLA typing in patients with multiple evanescent white dot syndrome (MEWDS). Ocul Immunol Inflamm, 1998. 6(1): p. 39-41.
- De Bats, F., et al., "En-face" spectral-domain optical coherence tomography findings in multiple evanescent white dot syndrome. J Ophthalmol, 2014. 2014: p. 928028.
- Pichi, F., et al., EN FACE OPTICAL COHERENCE TOMOGRAPHY AND OPTICAL COHERENCE TOMOGRAPHY ANGIOGRAPHY OF MULTIPLE EVANESCENT WHITE DOT SYNDROME: New Insights Into Pathogenesis. Retina, 2016. 36 Suppl 1: p. S178-S188.
- Marsiglia, M., et al., Expanded Clinical Spectrum of Multiple Evanescent White Dot Syndrome with Multimodal Imaging. Retina, 2016. 36(1): p. 64-74.
- Yannuzzi, N.A., et al., Swept-Source OCT Angiography Shows Sparing of the Choriocapillaris in Multiple Evanescent White Dot Syndrome. Ophthalmic Surg Lasers Imaging Retina, 2017. 48(1): p. 69-74.
- Pereira, F., et al., Swept-source OCT in patients with multiple evanescent white dot syndrome. J Ophthalmic Inflamm Infect, 2018. 8(1): p. 16.
- Furino, C., et al., Fundus autofluorescence and multiple evanescent white dot syndrome. Retina, 2009. 29(1): p. 60-3.
- Abou-Samra, A. and A.B. Tarabishy, Multiple Evanescent White Dot Syndrome Following Intradermal Influenza Vaccination. Ocul Immunol Inflamm, 2019. 27(4): p. 528-530.
- Yang, J.S., et al., Multiple evanescent white dot syndrome following rabies vaccination: a case report. BMC Ophthalmol, 2018. 18(1): p. 312.
- Stangos, A., et al., Multiple evanescent white dot syndrome following simultaneous hepatitis-A and yellow fever vaccination. Ocul Immunol Inflamm, 2006. 14(5): p. 301-4.
- Cohen, S.M., Multiple Evanescent White Dot Syndrome After Vaccination for Human Papilloma Virus and Meningococcus. J Pediatr Ophthalmol Strabismus, 2009.
- Jabs, D.A., et al., Classification criteria for multiple evanescent white dot syndrome. Am J Ophthalmol, 2021.
- Mantovani, A., et al., Multiple Evanescent White Dot Syndrome: A Multimodal Imaging Study of Foveal Granularity. Ocul Immunol Inflamm, 2019. 27(1): p. 141-147.
- Chen, K.C., et al., Foveal Exudate and Choroidal Neovascularization in Atypical Cases of Multiple Evanescent White Dot Syndrome. Retina, 2017. 37(11): p. 2025-2034.
- Cahuzac, A., et al., Multimodal imaging findings in 'hyper-early' stage MEWDS. Br J Ophthalmol, 2017. 101(10): p. 1381-1385.
- Onishi, A.C., et al., CHARACTERIZATION AND CORRELATION OF "JAMPOL DOTS" ON ADAPTIVE OPTICS WITH FOVEAL GRANULARITY ON CONVENTIONAL FUNDUS IMAGING. Retina, 2019. 39(2): p. 235-246.
- Feigl, B., A. Haas, and Y. El-Shabrawi, Multifocal ERG in multiple evanescent white dot syndrome. Graefes Arch Clin Exp Ophthalmol, 2002. 240(8): p. 615-21.
- Finn, A.P. and R.N. Khurana, Multiple evanescent white dot syndrome: Bilateral disease may be silent and asymmetric. Am J Ophthalmol Case Rep, 2021. 21: p. 101004.
- Sheng, Y., W. Sun, and Y.S. Gu, Spectral-domain optical coherence tomography dynamic changes and steroid response in multiple evanescent white dot syndrome. Int J Ophthalmol, 2017. 10(8): p. 1331-1333.
- Bosello, F., et al., Multiple evanescent white dot syndrome: clinical course and factors influencing visual acuity recovery. Br J Ophthalmol, 2020.
Suggested Citation Format
Motlagh M, Diel RJ, Sohn EH. Multiple Evanescent White Dot Syndrome - Case #1. EyeRounds.org. January 5, 2022. Available from https://eyerounds.org/cases/314-MEWDS.htm